

To evaluate the clinical features associated with adrenocortical hormone overexpression and familial cancer profiling as potential markers for early detection of adrenocortical tumors in children from South and Southeast Brazil.
MethodsThe clinical manifestations and anthropometric measurements of 103 children diagnosed with adrenocortical tumors were analyzed.
ResultsBetween 1982 and 2011, 69 girls and 34 boys diagnosed with adrenocortical tumors were followed‐up for a median time of 9.0 years (0–34 years). Signs of androgen overproduction alone (n=75) or associated with cortisol (n=18) were present in 90.3%. TP53 p.R337H mutation was found in 90.5% of patients. Stages I, II, III, and IV were observed in 45.6%, 27.2%, 19.4%, and 7.8% of patients, respectively. At diagnosis, there were no significant differences in height (p=0.92) and weight (p=0.22) among children with adrenocortical tumors, but children with virilization alone had significantly higher height‐for‐age Z‐scores (0.92±1.4) than children with hypercortisolism alone or combined (−0.32±1,8; p=0.03). The five‐year overall survival was 76.7% (SD±4.2). Patients with advanced‐stage disease had a significantly worse prognosis than those with limited disease (p<0.001). During follow‐up, ten of 55 p.R337H carrier parents developed cancer, whereas none of the 55 non‐carriers did.
ConclusionsSigns of adrenocortical hormone overproduction appear early, even in cases with early‐stage. These signs can be identified at the physical examination and anthropometric measurements. In southern Brazil, pediatric adrenocortical tumor is a sentinel cancer for detecting families with germline p.R337H mutation in TP53 gene.
Avaliar as manifestações clínicas da hiperexpressão de hormônios do córtex da adrenal e câncer familiar como marcadores para a detecção precoce de tumores adrenocorticais em crianças do Sul e Sudeste do Brasil.
Pacientes e métodosForam analisadas as manifestações clínicas e antropométricas de 103 crianças diagnosticadas com tumores adrenocorticais.
ResultadosEntre 1982 e 2011, 69 meninas e 34 meninos diagnosticados com tumores adrenocorticais foram acompanhados por um tempo mediano de nove anos (0‐34). Ao diagnóstico, sinais de virilização isolada (n=75) ou associada ao cortisol (n=18) estavam presentes em 90,3% dos pacientes; a mutação do gene TP53 p.R337H foi identificada em 90,5% dos pacientes. Os pacientes foram classificados em estádio I (45,6%), II (27,2%), III (19,4%) e IV (7,8%). Ao diagnóstico, não houve diferença significativa para as medidas de altura (p=0,92) e de peso (p=0,22) entre as crianças com tumores adrenocorticais, mas crianças com virilização tiveram escore‐Z mais elevado para a idade (0,92±1,4) do que aquelas com hipercortisolismo isolado ou combinado (−0,32±1,8; p=0,03). A sobrevida global de cinco anos foi de 76,7% (DP±4,2). Pacientes com estádios avançados tiveram pior prognóstico (p<0,001). Durante o seguimento, 10 dos 55 genitores portadores da p.R337H desenvolveram câncer, enquanto que nenhum caso ocorreu entre os 55 não portadores.
ConclusõesOs sinais de hiperprodução de hormônios adrenocorticais aparecem precocemente no desenvolvimento do tumor e podem ser identificados pelo exame físico e pelas medidas antropométricas na consulta pediátrica de rotina. O tumor adrenocortical pediátrico é sentinela para a detecção de câncer em famílias que segregam a mutação germinativa p.R337H do gene TP53.
Os tumores do córtex da glândula adrenal, comuns no Sul e Sudeste do Brasil, constituem um espectro de lesões com comportamento que varia de benigno a maligno, acometem principalmente crianças nos primeiros cinco anos de vida. Assumem importância significativa para a sobrevida, o diagnóstico e o tratamento precoces, uma vez que casos avançados podem ser altamente agressivos com alta mortalidade.
A incidência dos tumores do córtex da adrenal (TCA) no Sul e Sudeste do Brasil chega a 3,4 a 4,2 casos/milhão em crianças abaixo de 15 anos e representa uma frequência 18 vezes maior do que em outras regiões do Brasil e do mundo.1–3 Em mais de 90% dos casos estão associados à mutação germinativa do gene supressor de tumor TP53, localizada no éxon 10 do braço curto do cromossoma 17, denominada p.R337H, o que lhe atribui uma característica hereditária de predisposição a neoplasias, até o momento, exclusivas dessa população.4
Os resultados dos estudos de triagem neonatal, conduzidos na região de Campinas e no Estado do Paraná, mostraram que essa mutação está presente em cerca de um para 300 recém‐nascidos.5,6 Aparentemente, a mutação foi introduzida, como efeito fundador, nessa região brasileira no século XVIII, durante a fase de colonização do Brasil.7 A proteína codificada pelo gene TP53 é fundamental para manter a integridade genômica e mutações nesse gene levam a uma diminuição da atividade dessa proteína e, consequentemente, aumentam a probabilidade de câncer nos indivíduos carreadores.8 Devido à forte associação entre a mutação do TP53 e o TCA em crianças, esse tumor é considerado sentinela para a detecção da mutação TP53 p.R337H nos pacientes brasileiros.9 Outros fatores genéticos, epigenéticos e ambientais colaboram para estabelecer o risco nos indivíduos susceptíveis carreadores da mutação.10
As manifestações clínicas dos tumores adrenocorticais estão relacionadas à secreção aumentada de corticoesteroides (mineralocorticoides, glicorticoides e hormônios sexuais). O pediatra pode detectar os sinais e sintomas nas avaliações clínicas e antropométricas de rotina na puericultura, uma vez que cerca de 90% dos tumores são secretores; os não secretores são descobertos na avaliação clínica ou em exames ultrassonográficos para investigação diagnóstica do aumento do volume, da dor ou da massa abdominal.11
Neste artigo, os autores descrevem as principais características clínicas, epidemiológicas e biológicas do TCA na infância, com ênfase para a predisposição familiar ao câncer e estratégias para diagnóstico precoce, encaminhamento do paciente e orientação aos parentes sobre o risco de herdar a mutação.
MétodosElegibilidadeEntre dezembro de 19802 e dezembro de 2014, 137 pacientes abaixo de 23 anos foram diagnosticados com TCA no Centro Infantil Boldrini (CIB), referência em oncopediatria em Campinas/SP. Foram elegíveis para análise 103 pacientes e excluídos os pacientes com perda de seguimento (n=10), óbitos no perioperatório (n=4) e aqueles com menos de três anos do diagnóstico (n=20).
Acompanhamento longitudinalA partir de 2002, os pacientes do estudo seguiram o mesmo protocolo de acompanhamento clínico e laboratorial para diagnóstico da mutação entre os parentes, detecção precoce de recidiva, sequelas da terapia e ocorrência de câncer nos parentes.
As aferições do crescimento e desenvolvimento foram feitas nos retornos semestrais ou anuais, consideraram‐se os dados clínicos e as medidas antropométricas (peso, altura e índice de massa corpórea ‐ IMC). Foram usadas as curvas da Organização Mundial de Saúde como referência.12,13 Para análise do escore‐Z das medidas antropométricas, foi usado o programa Anthro Plus v.1.0.4 (WHO AnthroPlus software, versão 1.0.4, 2011).
Também foram elaborados heredogramas de três gerações das 55 famílias participantes do estudo familiar de câncer (programa Cyrillic v.2.1). Todos os pacientes receberam orientação genética e apoio psicológico.
Diagnóstico laboratorial e por imagensForam analisados os níveis séricos de testosterona total e livre, androstenediona, dehidroepiandrosterona, sulfato de dehidroepiadrosterona, aldosterona, cortisol, hormônio adrenocorticotrófico, progesterona e estradiol; raios X e tomografia de tórax, ultrassom e ressonância de abdome.
Ao diagnóstico, foram classificados de acordo com a manifestação clínica de excesso de hormônios corticoesteroides secretados pelo tumor: síndrome de virilização (androgênios), síndrome de Cushing (cortisol), síndrome combinada (virilização e Cushing) e assintomáticos (não secretores).
O volume do tumor foi mensurado no exame de imagem e o peso, aferido pela peça cirúrgica. O diagnóstico patológico seguiu a classificação de Weiss.14 Para detecção da mutação, foram feitos testes moleculares pelo método do PCR seguido de enzima de restrição e gel de agarose.4
Estadiamento clínico‐cirúrgicoOs pacientes foram estadiados de acordo com Sandrini et al.1:
ESTÁDIO I – Tumores<100g e<200cm3; completamente ressecados; níveis hormonais pós‐operatórios normais;
ESTÁDIO II – tumores ≥ 100g ou ≥ 200cm3; completamente ressecados; níveis hormonais pós‐operatórios normais;
ESTÁDIO III – tumores não ressecáveis; doença residual macro ou microscópica; ruptura do tumor; níveis hormonais altos após a cirurgia; envolvimento de linfonodos retroperitoneais;
ESTÁDIO IV – metástases a distância.
Aspectos do tratamentoPacientes com doença localizada foram submetidos à ressecção cirúrgica total do tumor. Os pacientes com tumores considerados inoperáveis ou com metástases a distância irressecáveis foram submetidos à biópsia para confirmação diagnóstica e receberam quimioterapia (QT) prévia à ressecção cirúrgica do tumor primário e das metástases. Os pacientes com doença avançada ou ruptura tumoral intraoperatória receberam QT intensiva com a combinação das drogas, cisplatina, etoposide, doxorrubicina associadas ao agente adrenocorticolítico mitotane (MTT) (o,p¿‐DDD).
Análise estatísticaForam feitas análises descritivas e de associação: apresentação de tabelas de frequência para variáveis categóricas; medidas de posição e dispersão para variáveis numéricas; associação entre as variáveis (teste qui‐quadrado ou teste exato de Fisher); para comparação de medidas numéricas entre grupos (teste de Mann‐Whitney ou Kruskal‐Wallis ou Anova com transformação por postos seguida pelo teste de Turkey); associação entre TCA e câncer (métodos Quase‐Likelihood ‐WQLS e teste Generalized Disequeibrium ‐ GDT); análise de sobrevida (teste de Kaplan‐Meier) e Log‐rank para comparação entre as curvas. O nível de significância foi de 5% e o programa Statistical Analysis System (SAS, software, versão 9.4, NC, EUA), versão 9.4, foi usado para análises.
Aspectos éticosO projeto de estudos dos TCA e da mutação germinativa nos pacientes e seus parentes foi aprovado pelos Comitês de Ética em Pesquisa do CIB e da Unicamp. Os sujeitos da pesquisa assinaram o termo de consentimento livre e esclarecido.
ResultadosAs características demográficas, clínicas e evolutivas dos pacientes estão apresentadas na tabela 1.
Características demográficas e clínicas de 103 pacientes com tumores adrenocorticais na infância e adolescência
| Características | n | % |
|---|---|---|
| Sexo | ||
| Feminino | 69 | 67,0 |
| Masculino | 34 | 33,0 |
| Cor | ||
| Amarelo | 1 | 1,0 |
| Branco | 88 | 89,8 |
| Negro | 4 | 4,1 |
| Pardo | 5 | 5,1 |
| Não informado | 5 | ‐ |
| Procedência por estado | ||
| São Paulo | 74 | 71,8 |
| Minas Gerais | 28 | 27,2 |
| Paraná | 1 | 1,0 |
| Faixa etária | ||
| ≤ 1ano | 50 | 48,5 |
| 2‐5 anos | 41 | 39,8 |
| 6‐10 | 5 | 4,9 |
| ≥ 11 anos | 7 | 6,8 |
| Mutação p.R337H do TP53 | ||
| Sim | 76 | 90,5 |
| Não | 8 | 9,5 |
| Não feito | 19 | ‐ |
| Síndrome endócrina | ||
| Virilização (androgênio) | 75 | 72,8 |
| Combinada (androgênio e cortisol) | 18 | 17,5 |
| Cushing (cortisol) | 3 | 2,9 |
| Assintomático (não secretor) | 7 | 6,8 |
| Pressão arterial | ||
| Elevada | 22 | 22,2 |
| Normal | 77 | 77,8 |
| Não informado | 4 | ‐ |
| Peso do tumor | ||
| ≤ 100 gramas | 52 | 56,5 |
| 100‐200 | 14 | 15,2 |
| ≥ 200 gramas | 26 | 28,3 |
| Não informado | 11 | ‐ |
| Volume do tumor | ||
| ≤ 200 cm3 | 65 | 67,7 |
| >200 cm3 | 31 | 32,3 |
| Não informado | 7 | ‐ |
| Estádio | ||
| I | 47 | 45,6 |
| II | 28 | 27,2 |
| III | 20 | 19,4 |
| IV | 8 | 7,8 |
| Terapia | ||
| Cirurgiaa | 72 | 70,0 |
| Cirurgia+QT+MTT | 18 | 17,4 |
| Cirurgia+MTT | 9 | 8,7 |
| Cirurgia+QT | 1 | 1,0 |
| QT+MTT | 3 | 2,9 |
| Resposta da terapia | ||
| Vivos | 79 | 76,7 |
| Óbitos | 24 | 23,3 |
| Causa do óbito | ||
| Complicações cirúrgicas | 2 | 8,3 |
| Progressão da doença | 16 | 66,7 |
| Encefalopatia hipertensiva | 3 | 12,5 |
| Encefalopatia por toxicidade | 3 | 12,5 |
O tempo mediano de história entre as manifestações clínicas e o diagnóstico foi de quatro meses (0,3‐36). O tempo mediano de seguimento dos pacientes foi de nove anos (variação, 0‐33,9). Não houve diferença significativa para a idade ao diagnóstico entre os sexos (p=0,27). Houve associação entre crianças com idade inferior a cinco anos e estádios I e II e entre crianças acima de 11 anos e estádios III e IV (p=0,02).
Houve correlação entre os sintomas ao diagnóstico e o estadiamento clínico para os pacientes com virilização e estádio I, pacientes com tumor combinado e estádios I e II e pacientes assintomáticos e estádios III e IV (p=0,01).
Houve correlação entre o peso dos tumores maiores do que 200cm3 e estádios avançados (III e IV), (p<0,0001).
Sete pacientes apresentaram associação com síndromes congênitas: Polipose Múltipla Familiar (2), Ectopia Pielocalicial (1), Imunodeficiência de Células NK (1), Duplo Arco Aórtico (1), Eritrodermia Variabilis (1), Síndrome de Beckwith Wiedmann (1); dos 84 pacientes testados, 76 (90,4%) foram carreadores da mutação germinativa TP53 p.R337H.
Dois pacientes portadores da mutação p.R337H apresentaram neuroblastoma sincrônico ao diagnóstico de TCA. Ocorreram dois casos de segunda neoplasia, 22 e 10 anos após o diagnóstico de TCA: uma paciente, não fumante, com adenocarcinoma de pulmão, e outra com osteossarcoma, respectivamente.
Os pacientes com doença localizada foram tratados com cirurgia para ressecção completa do tumor e os pacientes com doença avançada ou recidiva receberam terapia adjuvante (tabela 1).
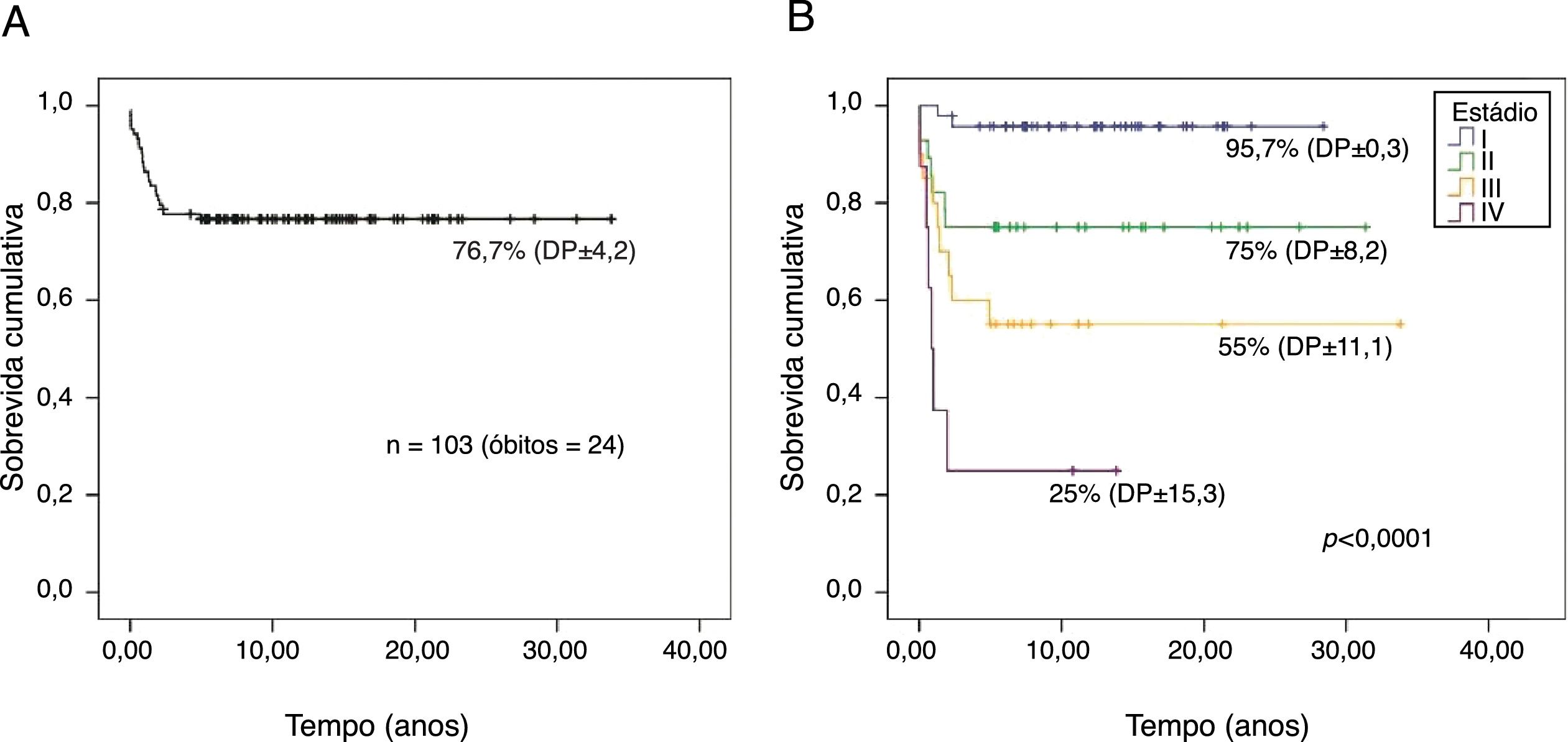
A probabilidade de sobrevida global dos 103 pacientes em acompanhamento por um tempo mediano foi de 26,2 anos (IC95%, 23,5‐28,9) e de 76,7% (DP±4,2) (fig. 1A). Houve diferença significativa para probabilidade de sobrevida global entre os estádios (p<0,001): estádio I, 95,7% (DP±0,3); II, 75% (DP±8,2), III, 55% (DP±11,1) e IV, 25% (DP±15,3) (fig. 1B).

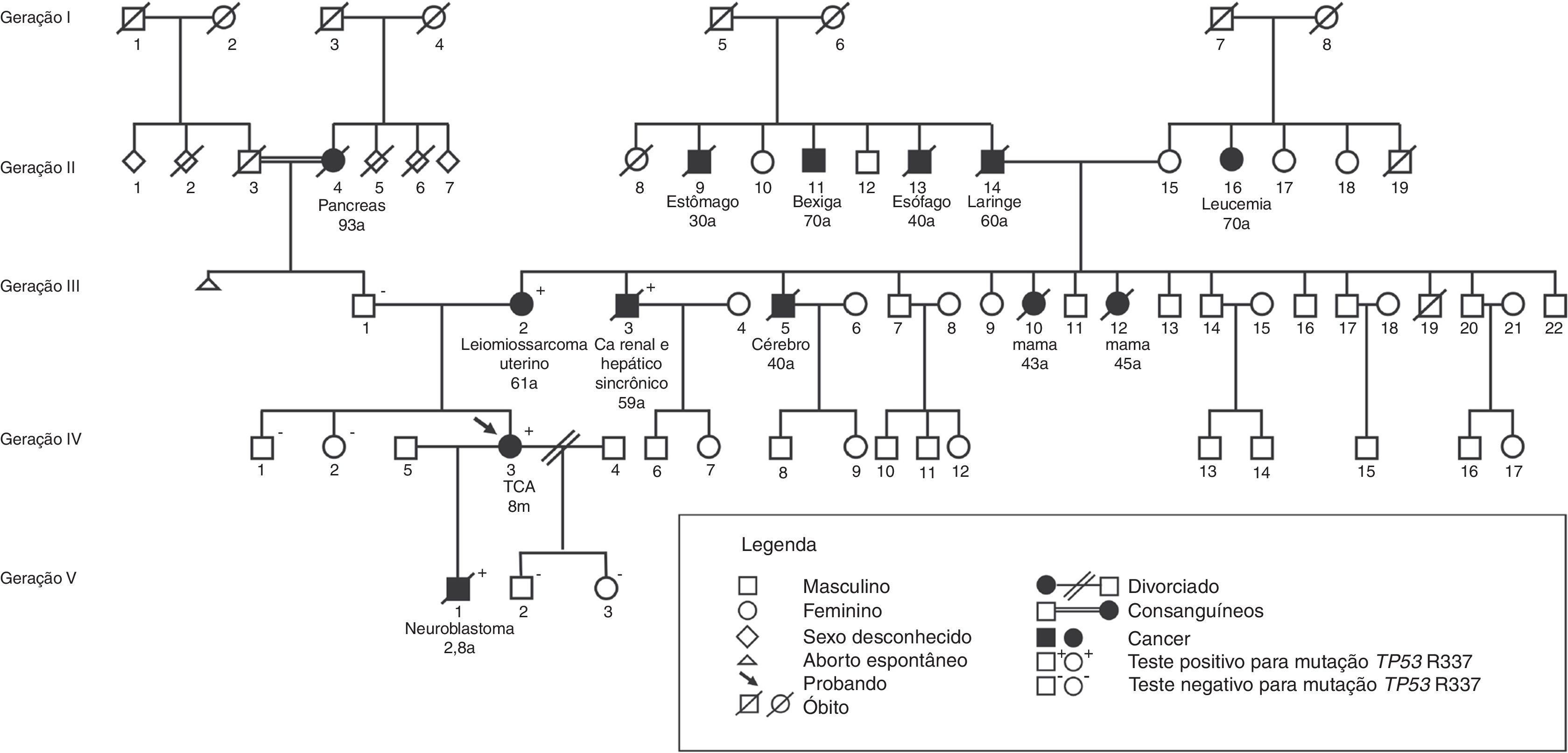
Dos 179 parentes de primeiro grau (genitores, irmãos, filhos) de 55 probandos, 175 foram testados para a mutação p.R337H. Desses, 13 de 81 carreadores e um de 94 não carreadores foram diagnosticados com câncer durante o período mediano de observação de 9,7 anos (3‐32) (p=0,001). Entre 33 mães carreadoras, três desenvolveram câncer de mama (aos 41, 43 e 44 anos), uma carcinoma de adrenal (47 anos) e uma leiomiossarcoma de útero (61 anos); entre os 22 pais carreadores, um desenvolveu tumor de sistema nervoso central (28 anos), um carcinoma de laringe (40 anos), um carcinoma de esôfago (46 anos), um adenocarcinoma de pulmão (51 anos, não fumante) e um câncer gástrico (62 anos); uma irmã de probando, carreadora, desenvolveu TCA aos 1,4 ano e um filho, carreador, neuroblastoma aos 2,8 anos; uma irmã, não carreadora, leucemia mieloide aguda (23 anos). A mediana dos genitores ao diagnóstico de câncer foi de 45 anos (28‐62).
A história familiar de câncer foi conhecida em 50 de 55 famílias da linhagem parental segregante (LPS) e 49 de 55da linhagem parental não segregante (LPNS) da mutação; 47/50 (94%) e 27/49 (55%) apresentaram história de câncer (p<0,001); múltiplas ocorrências de câncer em parentes de primeiro, segundo e terceiro graus foram observadas em 25 das 47 (53,2%) famílias LPS e quatro de 27 (14,8%) das famílias LPNS (p=0,001).
Os tipos de neoplasias mais comuns foram: câncer de mama 22/91 mulheres, 61 tumores gastrointestinais, 17 tumores de sistema nervoso central, 14 tumores de laringe em 198 neoplasias detectadas nas famílias que segregam a mutação.
Na figura 2, está exemplificado um heredograma de família com predisposição ao câncer.
Antropometria
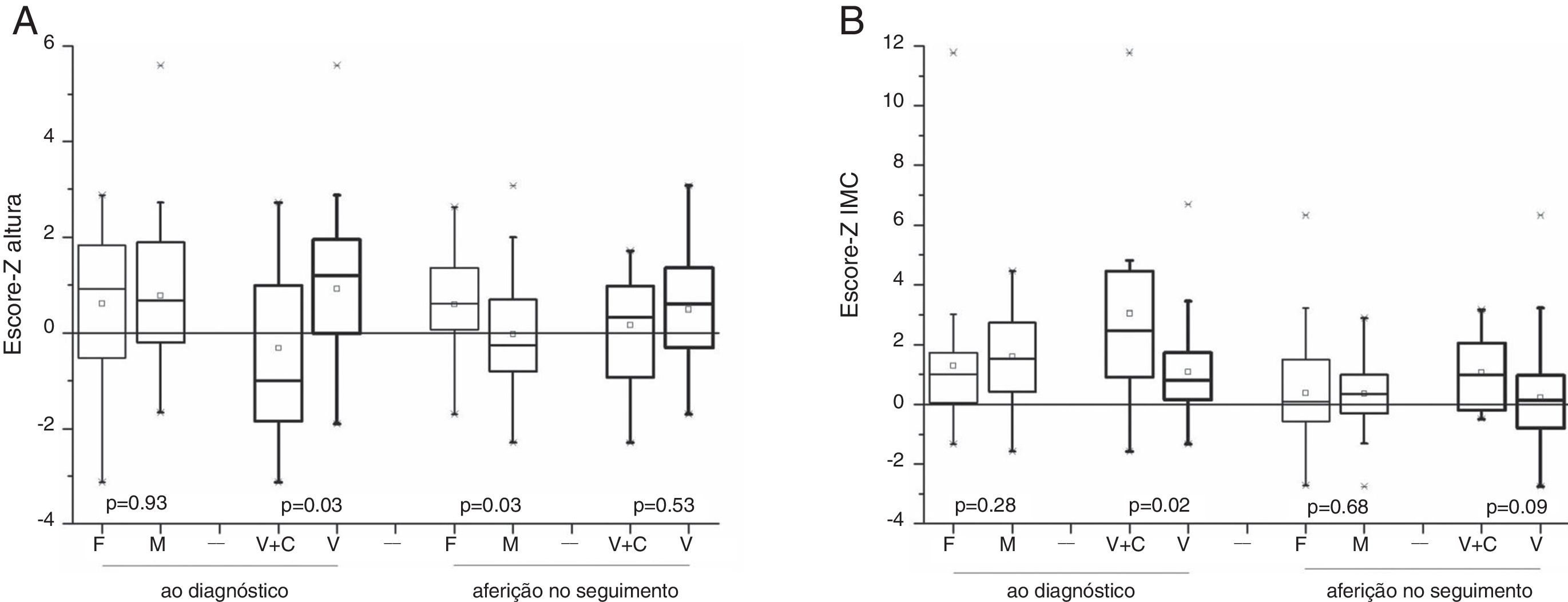
Os 64 pacientes que sobreviveram foram elegíveis para avaliação dos dados antropométricos. Ao diagnóstico, não houve diferença significativa para a média e mediana da idade (p=0,53), escore‐Z do peso (p=0,22), da altura (p=0,92) e do IMC (p=0,27) entre os sexos feminino e masculino. Os pacientes com sinais e sintomas de tumores secretores de androgênios apresentaram escore‐Z da altura acima da média para a população da mesma idade e sexo. Houve diferença estatística para o escore‐Z da altura (p=0,03) e do IMC (p=0,02) entre os pacientes com síndromes de virilização e combinada. Após a remissão da doença, nas consultas de seguimento, as análises antropométricas mostraram que não houve diferença estatística entre os sexos para o IMC (p=0,67), mas houve para o escore‐Z da altura maior para o sexo feminino (p=0,03); na comparação entre os pacientes com síndrome de virilização e combinada não houve diferença significativa para o escore‐Z da altura (p=0,52) e para o escore‐Z do IMC (p=0,08) (fig. 3).
 e do IMC (B) entre os sexos e entre as síndromes endócrinas (virilização e combinada), em dois momentos de avaliação: ao diagnóstico e após a terapia de 64 sobreviventes (45 do sexo feminino e 19 do masculino) de TCA na infância.")
A análise do catch down da altura para os pacientes com virilização mostrou maior queda no escore‐Z para o sexo masculino (p=0,03).
DiscussãoNeste estudo, os autores confirmaram que o diagnóstico precoce de crianças portadoras da mutação p.R337H e TCA está associado com a alta curabilidade sem prejuízo à saúde em longo prazo. Cirurgia com ressecção completa do tumor é suficiente para curar aproximadamente 80% das crianças com tumores pequenos. Contudo, quando o diagnóstico é tardio, os tumores são mais agressivos e os pacientes recebem tratamento adjuvante com quimioterapia intensiva e mitotane, o que compromete o prognóstico e a sobrevida.
Os resultados demonstraram que os sinais e sintomas iniciais do TCA podem ser detectados no acompanhamento de puericultura, uma vez que 86% dos casos ocorrem antes dos cinco anos e 93% dos pacientes apresentam distúrbios do crescimento somático ou caracteres sexuais inapropriados para a idade.15,16 Deve ser ressaltado que a secreção anormal de esteroides ocorre precocemente durante o desenvolvimento do tumor, por isso as manifestações clínicas sutis representam os primeiros sinais da doença.
A aceleração do crescimento em altura pode ser o primeiro sinal de secreção elevada de andrógenos e o ganho rápido de peso, de secreção de cortisol; neste estudo, 75% das crianças estavam acima do percentil 75 para peso e altura. Para as crianças com tumores não secretores, o escore‐Z para peso e altura ficou abaixo da média, provavelmente pela perda secundária ao catabolismo do tumor, uma vez que o diagnóstico foi mais tardio, com doença avançada.
A maioria dos casos apresenta‐se como pseudopuberdade precoce com sinais de virilização ou como síndrome de Cushing, isolada ou associada, facilmente confundidos com hiperplasia da glândula adrenal ou puberdade precoce central.17,18 Destacamos que, particularmente em crianças das regiões Sul e Sudeste do Brasil, a possibilidade de TCA deve ser a primeira hipótese e deve ser feito de imediato o exame de imagem, ultrassom, ou preferencialmente ressonância magnética (RNM) de abdome, uma vez que o atraso no diagnóstico compromete o prognóstico e a sobrevida.19 Não é indicada tomografia computadorizada pelo risco de exposição à irradiação. Devido às possíveis dificuldades para feitura de RNM, sugere‐se o exame de ultrassom de abdome, com especial atenção às glândulas adrenais, e a coleta dos hormônios (androgênios e cortisol), além da investigação da história familiar de câncer. A presença de virilização antes dos quatro anos, hipercortisolismo antes dos 10 anos, desproporção entre sinais de virilização e volume testicular em meninos e presença de sinais de duas cadeias de esteroides é altamente sugestiva de TCA.16 Nesses casos e/ou presença de massa em adrenal, a criança deve ser encaminhada ao centro de referência com urgência.
A hipertensão arterial foi observada ao diagnóstico em aproximadamente 22% dos pacientes e, na maioria, associada à produção de cortisol. A hipertensão foi mais comum nos pacientes com síndrome de Cushing e combinada. Entretanto, 7% dos pacientes com virilização ou tumor não secretor apresentaram hipertensão, provavelmente devido à compressão da artéria renal pelo tumor ou por aumento da produção de aldosterona.11
Outros sinais e sintomas relacionados aos tumores secretores que podem estar presentes são: pubarca ou pelos axilares prematuros (92%), clitoromegalia (92%), falomegalia (91%), hirsutismo (62%), aumento do volume ou massa palpável no abdome (55%), acne (47%), pletora facial (42%), fácies em lua cheia (35%), aumento do timbre da voz (32%); a telarca precoce não é um sinal comum, pois os tumores produtores de estrogênios são raros; a hipertensão secundária à secreção de aldosterona, geralmente, é uma manifestação associada à virilização ou ao hipercortisolismo.11,20,21
Neste estudo, os TCA não secretores representaram 6,8% dos casos e foram diagnosticados tardiamente por exames de imagem, com doença avançada e prognóstico reservado.
A exposição prolongada aos hormônios androgênicos acelera a idade óssea e o desenvolvimento, algumas vezes leva à puberdade precoce com necessidade de bloqueio hormonal.17,20 Na nossa casuística, tivemos três meninas com necessidade de bloqueio puberal aos seis e sete anos.
O prognóstico do TCA está associado ao estadiamento da doença ao diagnóstico. Neste estudo, a probabilidade de sobrevida para os pacientes com doença localizada, passível de ressecção cirúrgica, foi acima de 75%. O mitotane (o‐p’DDD), agente adrenocorticolítico, tem sido usado como adjuvante à cirurgia, associado ou não à quimioterapia convencional, porém seu real valor precisa ser comprovado. Além disso, o mitotane tem efeitos colaterais importantes.22 A radioterapia não é preconizada, pois, em mais que 90% dos casos no Sul do Brasil, o carcinoma de adrenal está associado a mutações do TP53, pode predispor a neoplasias secundárias na área irradiada.21
A mutação TP53 p.R337H está associada a outras neoplasias na infância e na fase adulta. O grupo desta pesquisa demonstrou a associação da p.R337H com outros tumores pediátricos entre as crianças tratadas no CIB, revelou que, além do TCA, a p.R337H está fortemente associada ao carcinoma do plexo coroide (69%), osteossarcoma (7%) e neuroblastoma (8,4%) e em 3/123 mulheres com câncer de mama na Região Sudeste do Brasil.23–25
Em todos os casos de pacientes portadores da p.R337H a herança foi comprovada pela presença da mutação em um dos genitores, corroborou a observação que até o momento, no Sul do Brasil, não são relatados casos de mutações de novo. Em 2001, Ribeiro et al. descreveram, pela primeira vez, a mutação missense no éxon 10 do gene TP53 que originou a variante da proteína com substituição de uma arginina por uma histidina denominada p.R337H e comprovaram a associação dessa mutação com o TCA.4 Outros autores têm relatado a associação dessa mutação com famílias portadoras de síndromes de predisposição familiar ao câncer.26
No estudo prospectivo de 55 famílias de crianças tratadas de TCA e portadoras da mutação, encontramos 13 novos casos de câncer entre os 81 parentes de primeiro grau testados carreadores e um caso entre 94 parentes não carreadores. Na literatura, outras mutações não p.R337H do TP53 estão associadas à síndrome de câncer familiar, a clássica síndrome de Li‐Fraumeni e suas variantes Li‐Fraumeni Like (OMIM #151623), porém com espectro de tumores diferentes (sarcomas, carcinoma de mama, tumores de sistema nervoso central e leucemia) do que os encontrados nos parentes deste estudo e idade de ocorrência de câncer em indivíduos mais jovens.27–29
Os autores orientam incluir na anamnese a história familiar de câncer e o heredograma de três gerações para alertar sobre a presença da mutação, uma vez que, por ser hereditária, os indivíduos na linhagem segregante estão sob risco de desenvolver câncer. A presença de câncer em mais de um parente de primeiro e segundo graus, ou tumores múltiplos no mesmo indivíduo ou câncer em indivíduos jovens, fala a favor de predisposição familiar.
Os autores desta pesquisa demonstraram a alta frequência de TCA em um centro de referência do Sudeste do Brasil, associado à mutação hereditária do TP53 p.R337H em mais de 90% dos casos; a maioria dos casos pode ser detectada precocemente pelos sinais e sintomas de secreção elevada de corticoesteroides; a sobrevida está relacionada ao diagnóstico precoce e à doença localizada; o diagnóstico de TCA numa criança representa um alerta para predisposição familiar ao câncer.
O pediatra, na atenção primária de saúde, é o elo fundamental entre a suspeita diagnóstica e o tratamento adequado. Cabe aos centros de referência desenvolver protocolos terapêuticos e estratégias que possibilitem ao pediatra novas formas de prevenção e medidas preditivas para crianças e parentes sob risco de desenvolver câncer.
FinanciamentoProjeto sob coordenação do professor José Andrés Yunes com bolsas do Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) n° 401991/2010 e Fundação Capes, Ministério da Educação, Procad n° 247/2007 para análises de biologia molecular.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
A Gisele Miniussi de Assis, gerente de dados do Programa de Estudos dos Tumores Adrenocorticais na Infância e do Protocolo de Investigação ARAR0332 do Children's Oncology Group, desenvolvidos no Centro Infantil Boldrini e na Unicamp. A Cleide Aparecida Moreira Silva, estatística do Serviço de Estatística da Faculdade de Ciências Médicas da Unicamp.
Como citar este artigo: Mastellaro MJ, Ribeiro RC, Oliveira‐Filho AG, Seidinger AL, Cardinalli IA, Miranda EC, et al. Adrenocortical tumors associated with the TP53 p.R337H germline mutation can be identified during child‐care consultations. J Pediatr (Rio J). 2018;84:432–9.





