


The aim of the report is to describe the main immunodeficiencies with syndromic characteristics according to the new classification of Inborn Errors of Immunity.
Data sourceThe data search was centered on the PubMed platform on review studies, meta-analyses, systematic reviews, case reports and a randomized study published in the last 10 years that allowed the characterization of the several immunological defects included in this group.
Data synthesisImmunodeficiencies with syndromic characteristics include 65 immunological defects in 9 subgroups. The diversity of clinical manifestations is observed in each described disease and may appear early or later, with variable severity. Congenital thrombocytopenia, syndromes with DNA repair defect, immuno-osseous dysplasias, thymic defects, Hyper IgE Syndrome, anhidrotic ectodermal dysplasia with immunodeficiency and purine nucleoside phosphorylase deficiency were addressed.
ConclusionsImmunological defects can present with very different characteristics; however, the occurrence of infectious processes, autoimmune disorders and progression to malignancy may suggest diagnostic research. In the case of diseases with gene mutations, family history is of utmost importance.
The recognition of new immunological defects that translate into immune dysregulation or autoinflammation and, more recently, bone marrow failures has caused the name of “Immunological defects” to be changed to Inborn Errors of Immunity. Since the last classification, 65 new conditions have been included, totaling 430 phenotypes. Therefore, these diseases are classified into 10 groups and the group of Combined Immunodeficiency Syndromes comprises diseases with the most diverse immunological disorders. These include nine tables and 63 genes.1 (Table 1).
Combined immunodeficiencies associated with syndromes.
| Syndromes | Examples |
|---|---|
| Congenital thrombocytopenia | Wiskott-Aldrich syndrome |
| Other DNA repair defects | Ataxia-telangiectasia |
| Immune-osseous dysplasia | Cartilage-hair hypoplasia |
| Thymic defects | Di George syndromes/Velocardiofacial/ Cr.22q11.2deletion |
| Hyper-IgE Syndrome | |
| Defects of Vitamin B12 and folate metabolism | |
| Anhidrotic ectodermal dysplasia with immune deficiency | NEMO |
| Others | Purine nucleoside phosphorylase deficiencyCalcium Channel Defects |
In the European Society for Immunodeficiency (ESID) Registry and the Latin American Society for Immunodeficiencies (LASID)Registry, it was found that 15.6% and 13.5% of Primary Immunodeficiencies (name used in the publication), respectively, corresponded to the group of immunological defects associated with syndromes or also included with the title “well defined”2 (LASID, 2020, unpublished data). Regarding frequency, therefore, they follow immunodeficiencies predominantly of antibodies.
Aiming to summarize the main clinical and immunological characteristics, a search for reviews on the topic was carried out that would allow the presentation of the most recently acquired knowledge. Due to the diversity of phenotypes of the diseases included, they will be shown separately.
Congenital thrombocytopeniaWiskott-Aldrich syndrome (OMIM 301000) has an incidence of approximately one to four cases per 1,000,000 male live births. The affected gene is located on the short arm of the X chromosome (Xp11.22 – p11.23),1 expressed only in hematopoietic cells. Female carriers are usually asymptomatic.3 This gene encodes the WASp protein,4 which coordinates the organization of actin filaments in response to cell signaling events in the cytoskeleton. Thus, it was demonstrated that defects in WASp function impair the processes in myeloid and lymphoid lineage cells, including cell adhesion and migration, phagocytosis, immunological synapse structure, autophagy, and inflammation. The pathogenesis of the platelet defect remains partially understood. Megakaryocytic dysfunction is suspected to be a hypothesis, leading to the formation of small and defective platelets, associated with their increased destruction in the spleen.5,6
The complete absence of the protein leads to a marked function defect in multiple hematopoietic cell lines, resulting in thrombocytopenia with small platelets and progressive lymphopenia with abnormal lymphoid and myeloid functions, which is classified as the “classic” form of the disease.1,6 The milder spectrums of the disease are X-linked thrombocytopenia (XLT) and X-linked neutropenia.3
In its classic form, Wiskott-Aldrich syndrome appears in the first year of life with a triad of recurrent infections, eczema and microthrombocytopenia, with petechiae, hematomas, spontaneous or prolonged bleeding. Occasionally, mild to moderate thrombocytopenia can manifest in a more advanced stage of childhood, mimicking the clinical picture of Idiopathic Thrombocytopenic Purpura (ITP), albeit without response to oral steroids.4 Sinopulmonary infections are the most common infectious complications before the diagnosis, including otitis media and pneumonia. Severe infections can occur, such as sepsis and meningitis. These patients have increased susceptibility to opportunistic infections by organisms such as Pneumocystis jirovecii. They can develop severe and widespread forms of viral infections, mainly by the herpes simplex I or II virus and the varicellazoster virus, in addition to fungal infections caused by invasive yeast.1
This syndrome also occurs with autoimmune manifestations, which may include autoimmune hemolytic anemia, autoimmune neutropenia, autoimmune vasculitis, IgA nephropathy, arthritis and inflammatory bowel disease. The development of malignant diseases, mainly lymphomas, has also been described.3
X-linked thrombocytopenia (XLT), the mildest spectrum of the syndrome, has a similar bleeding phenotype, but without other significant clinical features.4
For the diagnosis of Wiskott-Aldrich syndrome, the cytoplasmic quantification of the WASp protein by flow cytometry is an efficient and rapid test.5 Genetic analysis is the gold standard for the diagnostic confirmation and plays an important role in patient management decisions and family screening.4 IgG, IgA and IgM levels may be low or high as a result of altered antibody-mediated immunity. IgE levels are usually high. The vaccine response to protein antigens is generally conserved; however, responses to polysaccharide antigens and isohemagglutinins are unsatisfactory.4,6
Hematopoietic stem cell transplantation is the treatment of choice for the classic form of the syndrome and shows better results within the first two years of life.1,4 The survival of children submitted to early hematopoietic stem cell transplantation is, however, excellent, with survival rates above 97%. Gene therapy is also a therapeutic option restricted to patients without a fully compatible bone marrow donor.3
Syndromes related to DNA repair defectsAtaxia-telangiectasia (A-T)Ataxia-telangiectasia (AT) (OMIM 607585), as well as Nijmegen breakage syndrome (NBS) (OMIM 602667) and Bloom's syndrome (BS) (OMIM 604610), is transmitted by autosomal recessive inheritance with a high rate of parental consanguinity.7 It has an estimated prevalence of 1:40 000 to 1:100000 worldwide.7 It is caused by mutations in the encoding of the ataxia-telangiectasia mutated (ATM) gene. The primary role of nuclear ATM is the coordination of cell signaling pathways in response to double stranded DNA breaks, oxidative stress and the cell cycle checkpoint. Most ATM mutations lead to a truncated protein, while some variants at the splicing site cause milder phenotypes. The ATM also plays an important role in hematopoietic cells and neurons. Moreover, the ATM adjusts the functions of organelles such as mitochondria and peroxisomes, in addition to regulating angiogenesis and glucose metabolism.8
AT patients have progressive cerebellar ataxia, oculocutaneous telangiectasia, variable immunodeficiency, radiosensitivity, predisposition to malignancy and an increase in metabolic diseases. This congenital disease has phenotypic heterogeneity, is progressively established, and symptom severity varies in different patients based on the severity of mutations and disease evolution.9 Oculomotor apraxia often precedes the development of telangiectasias, which are established between 3 and 5 years of age.7 Mental retardation is not a feature of AT, although some older patients experience severe short-term memory loss.10 Due to several neurological manifestations, patients are initially often referred to neurology. Other abnormalities, such as failure to thrive, low pubertal development, insulin-resistant diabetes, gonadal atrophy, pulmonary disease, skin abnormality, and cardiovascular disease have also been reported in AT patients.10 AT patients and their cultured cells are uncommonly sensitive to x-rays; therefore, imaging tests in these patients should be cautiously performed, due to the risk of malignancy.7 Considering the aforementioned findings, associated with a high level of serum alpha-fetoprotein and immunological deficiencies, they suggest the diagnosis of ataxia-telangiectasia, and these pathogenic mutations in the gene (ATM) prove the clinical diagnosis.8
The immunological defects described are: decreased or absent serum levels of IgG2 and IgA in 80% and 60% of patients, respectively; decreased IgE levels; decreased or normal IgM levels; peripheral lymphopenia, even in the early stages of the disease and thymichypoplasia. Some AT patients can be identified in the neonatal screening for severe combined immunodeficiency (SCID) with very low levels of T-Cell Receptor Excision Circles (TREC).7
Mild phenotypes were defined as those with late onset, with no ataxia on presentation or with ataxia not being the dominant feature, or with slower progression, prolonged life compared to most patients with AT and decreased levels of chromosomal instability and cell radiosensitivity.7
Unfortunately, no curative therapy is available for ataxia-telangiectasia. Given the complexity and severity of the disorder, patients should receive optimal symptomatic treatment in the context of a dedicated and experienced multidisciplinary team. The prognosis is poor and the time of survival, at the moment, is approximately 25 years. The most common causes of death in these patients are chronic pulmonary disease and malignancy, such as leukemia. In general, lymphomas in patients with AT tend to originate in B cells, whereas leukemias tend to be of T cells.9
Nijmegen breakage syndrome (NBS)Nijmegen breakage syndrome (NBS) (OMIM 602667) has been described in many patients with restricted geographic origin (Slavs and, particularly, individuals of Polish or Czech descent) and who carry a common founder mutation, 657del5 in exon 6 of NBN gene (formerly NBS1).11 It is characterized by dysmorphic facial features, which become accentuated with advancing age. They have a prominent forehead and recessed jaw. Intrauterine growth retardation is usually present and patients have marked microcephaly at birth. Other characteristics include short stature, congenital skeletal (clinodactyly, syndactyly) andrenal abnormalities and mild non-progressive mental retardation.
Premature ovarian failure is reported in women. Sinopulmonary infections are common, as well as malignancies, particularly lymphomas of B-lymphocyte lineage cells, and autoimmune manifestations, which include pulmonary granuloma and interstitial lymphocytic lung disease. Cell and humoral immune deficiency is widely reported, with variable clinical expression intensity. The occurrence of hypogammaglobulinemia is associated with chronic recurrent infections of the respiratory tract. This clinical spectrum can lead to the development of bronchiectasis that requires immunoglobulin replacement. Opportunistic infections are rare and there is generally no correlation between the degree of cell deficiency and infection severity. Some patients with Nijmegen's breakage syndrome can also be identified in the neonatal screening for SCID with very low levels of TRECs.8
Malignancy remains the most significant risk for patients with NBS, with most tumors originating from the lympho-reticular system. The mean age of malignancy onset is around 10 years old. The role and timing of preventive hematopoietic stem cell (HSCT) transplantation is not yet fully established.
Bloom syndrome (BS)Bloom syndrome (BS) (OMIM 604610) has an estimated prevalence of 2 in 1,000,000, with approximately 300 cases reported worldwide.11 It is characterized by abnormal pre- and postnatal growth, feeding difficulties in childhood, sensitivity to sunlight, immune deficiency, increased risk of diabetes, insulin resistance and malignancy.12 The facial features of individuals with BS are variable and can be indistinguishable from their peers of the same age. They may have a long, narrow face, underdeveloped malar area and retrognathia or micrognathia. The nose and/or ears are often prominent. Despite their very small head circumference, most affected individuals have normal intellectual capacity. The women may be fertile, but often experience early menopause, and men tend to have infertility.12 The clinical diagnosis can be confirmed by cytogenetic analysis that identifies an increased number of sister chromatid exchange levels. The molecular confirmation of BS identifies biallelic mutations of BLM.12 The mean survival reported for patients is 27 years, but the treatment of complications and malignancy can greatly extend the survival of these patients.11 Heterozygous carriers may have an increased risk of cancer.
Immuno-osseous dysplasiasCartilage-Hair hypoplasia (CHH)CHH (OMIM 250250) has an autosomal recessive inheritance and its prevalence was estimated at 1:23,000 live births.1,13 Patients have skeletal dysplasia, short stature, hypotrichosis, varying degree of immunological dysfunction, increased incidence of anemia, Hirschsprung's disease and malignancy.13,14
Infections in CHH patients commonly include recurrent episodes of otitis media, sinusitis and pneumonia, caused by common pathogens such as Haemophilus influenzae, Moraxella catarrhalis and Streptococcus pneumoniae. Infections by Pneumocystis jiroveci, Aspergillus spp and herpes viruses (particularly varicella-zoster virus) have also been described in severe cases. Not all patients have recurrent or severe infections, although cell immunity is clearly impaired, confirmed through low lymphocyte subpopulation counts, as well as abnormal T-cell proliferation. Interestingly, CHH patients with bronchiectasis have a higher T-cell count and higher levels of IgG. Deficiency of specific antibodies has been reported in most evaluated patients and can be a marker of a more severe disease course.14
In addition to bronchiectasis, high rates of fibrosis, as well as fatal cases of pulmonary emphysema have been reported. Given the high prevalence of clinically relevant pulmonary alterations, pulmonary magnetic resonance imaging should be performed on all adults and symptomatic children with CHH.14
Up to 11% of CHH patients develop autoimmune diseases, such as: enteropathy, hemolytic anemia, hypoparathyroidism, hypo or hyperthyroidism, idiopathic thrombocytopenic purpura, juvenile rheumatoid arthritis, multifocal motoraxonal neuropathy, narcolepsy, neutropenia, and psoriasis. Immune dysregulation in CHH can also manifest with hepatosplenomegaly, severe eczema and inflammatory granulomatous skin lesions. Granulomas in CHH can be positive for the rubella virus of the vaccine strain and can be extremely difficult to treat. It is important to emphasize that cutaneous granulomas in individuals with CHH may also represent a reaction to infection by the Epstein-Barr virus.
Patients with CHH are at increased risk of malignancy, especially lymphoma and basal cell carcinoma. The most common malignancy is non-Hodgkin's lymphoma. The distribution of skin cancer in these individuals is directly related to sun exposure. The pathogenesis of malignancy in CHH is multifactorial; however, it has also been associated with the Epstein-Barr virus. There is also difficulty in controlling the human papillomavirus (HPV) infection.14
CHH is caused by variants in the RMRP gene, which encodes the untranslated RNA molecule of the mitochondrial RNA processing endoribonuclease, which participates, for instance, in the cell cycle regulation and telomeremaintenance. Neonatal screening for SCID may possibly have a prognostic value in CHH. Regular monitoring by a multidisciplinary team should be implemented to treat immune dysfunction in all patients with CHH, and also in asymptomatic cases. Hematopoietic stem cell transplantation can cure the immune dysfunction, but its benefits in mildly symptomatic patients with CHH remain debatable.13
Schimke immuno-osseous dysplasia (SIOD)SIOD is an autosomal recessive multisystem disorder, with an estimated incidence in North America of 1:1,000,000 to 1:3,000,000 live births.15 Spondyloepiphyseal dysplasia occurs, resulting in short stature, nephropathy and T-cell deficiency.16 Radiographic manifestations include ovoid and slightly flattened vertebral bodies, small deformed femoral epiphyses, dysplastic and shallow acetabular fossae, with an average adult height of 98.5−157cm. SIOD involves a spectrum that varies from an infantile or severe form, with death early in life, to a juvenile or lighter form, of late onset, with survival to adulthood if kidney disease is adequately treated.15 Phenotypic heterogeneity and variable expressivity suggest that SIOD is modified by factors such as environment, epigenetics and oligogenic inheritance. Therefore, the follow-up of these patients must be performed by a multidisciplinary team with a focus on nephrological, hematological and immunological alterations.16
Thymic defectsDiGeorge syndromeDiGeorge's syndrome (OMIM 188400) is one of the clinical spectrums of chromosome 22q11.2 deletion syndrome,17 with an incidence of 1:4,000 live births, although the actual frequency may be even higher. The main features of this syndrome are cardiac, thymic and parathyroid disorders, structures that have the same embryological origin.18
Thymic hypoplasia is the main feature of the syndrome, and as a consequence, T-cell alterations. Some patients have a completely normal T-lymphocyte count (depending on age), while others have greater impairment.19 B-cell dysfunction can also occur, resulting in low levels of immunoglobulins.18,19
Autoimmune diseases are also part of DiGeorge's syndrome, such as autoimmune cytopenias and idiopathic arthritis.17,19 Extreme lymphopenia can lead to a Th2 distortion, strongly associated with atopy.19 Most cardiovascular abnormalities are conotruncal defects, such as tetralogy of Fallot, truncus arteriosus, interrupted aortic arch, and ventricular septal defect.18
Hypoparathyroidism is an important feature of the syndrome and can lead to tetany, seizures, feeding difficulties, stridor and fatigue.18,19 Other alterations, such as hypothyroidism, short stature, delayed motor and language development, anxiety, autistic spectrum disorder and attention deficit may also be present.18
For an early diagnosis, the TREC test is performed shortly after birth, which is a screening test for severe immunodeficiencies related to T-cell lymphopenia.17,18 T-cell count in these patients is sometimes normal early in life, and cell depletion may occur over the months, with a subsequent decrease in the number of T lymphocytes. Periodic monitoring of these patients is prudent, given the lack of biomarkers for the evolution of immune deficiency17 The obstetric ultrasound can be very useful in identifying cardiac and thymic abnormalities.18
Heart defects are the leading cause of death in DiGeorge's syndrome.18 Prophylactic antibiotics or human immunoglobulin replacement therapy can be used to prevent infections.18 The risk of administering live virus vaccines to these patients is low, with the exception of patients with thymic aplasia and/or very low T-cell count.17 Thymus or fully compatible T-cell transplantation may be necessary in patients with thymic aplasia; however, it is estimated that only 1% of patients have the most severe disease type.17
CHARGE syndromeCHARGE syndrome (CS) (OMIM 214800) has an incidence of 1 in 10,000 to 15,000 live births. It has an autosomal dominant inheritance in 95% of cases, due to a heterozygous mutation in the CHD7 gene, located in the long arm of chromosome 8 (8q12). It is a de novo mutation, but the parents of a child with this syndrome have a 1 to 3% chance of recurrence in the next child.20 The CHD7 gene is essential in intrauterine life for the migration of neural crest cells, which differentiate into a variety of tissues, including cardiac, genitourinary, neural, craniofacial, thymic and parathyroid tissue.21
CHARGE is an acronym that describes the main components of this syndrome: coloboma of the eye, heart defect, choanal atresia or stenosis, growth and development retardation, genitourinary abnormalities and ear abnormalities.21 The patient clinical phenotype may include facial asymmetry (due to facial nerve palsy), cup ear deformity with decreased cartilage and triangular concha, cleft lip, square-shaped face with prominent forehead and eye alterations.21,22 Cardiac malformations are present in 65–75% of patients with this syndrome, manifesting mainly as conotruncal defects (deformities in the aortic arch, Tetralogy of Fallot and right ventricle double outlet) and atrioventricular septal defect. Hypogonadotropic hypogonadism is also common, with micropenis, cryptorchidism and clitoral reduction. The kidneys can be horseshoe-shaped, ectopic or dysgenetic. These patients usually have normal weight and length at birth, but by the end of childhood they already show significant growth deficit. Cognitive ability is impaired, as well as communication and language.19 Conductive and/or sensorineural hearing loss can occur in CHARGE syndrome. The most frequent abnormality of the inner ear is the absence of lateral semicircular canals, but vestibular (all semicircular canals) and cochlear dysplasia can also occur, resulting in variable spectrums of sensorineural hearing loss. Vestibular hypoactivity leads to an alteration in balance, which in turn causes delayed gait.19 Conductive hearing loss can be caused by dysfunction in the auditory tube or by dysplastic or absent middle ear ossicles.20,21
The mnemonic term CHARGE does not include all the features of the syndrome. More than 90% of affected individuals have dysfunction of the cranial nerves (CN), manifesting as absent or reduced sense of smell (CN I), weak chewing and swallowing (CN V), facial paralysis (CN VII), sensorineural hearing loss (CN VIII), vestibular balance problems (CN VIII) and swallowing problems (CN IX and X). Visual and hearing impairments, as well as intellectual disabilities are prevalent and can range from mild to severe.20
Immunodeficiency in CHARGE syndrome is rare and occurs mainly due to impaired thymic development, impairing the production of functional T cells. Immunodeficiency severity is related to the degree of inadequate thymic development. Complete thymic aplasia is rare and causes severe immunodeficiency with partial or total absence of T cells and abnormal B-cell function with associated hypogammaglobulinemia.23
Partial thymic aplasia can result in several clinical phenotypes: no detectable defect; slight reduction in T cells with no clinical consequence; more significant reduction in T cells with impaired B-lymphocyte function, resulting in recurrent infections that will require antibiotic prophylaxis or human immunoglobulin infusion. Isolated defects in B lymphocytes or antibodies are rarely described in CHARGE syndrome.23
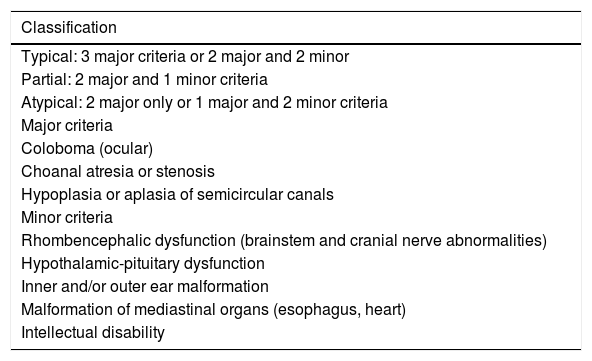
The clinical diagnosis is attained by the major and minor criteria modified by Verloes et al. (2005)22 (Table 2). A complete clinical evaluation of an individual with suspected CS should include echocardiography, renal ultrasonography, chest ultrasound, computed tomography and magnetic resonance imaging of the skull, audiometry, cranial nerve testing, deglutition study, nasal endoscopy and fundoscopy, complete blood count (CBC), immunoglobulin levels, and lymphocyte immunophenotyping, among other tests, individualizing each case. The diagnosis is confirmed by genetic analysis.20 SANGER sequencing of the CHD7 gene will detect CHARGE-causing mutations in 90 to 95% of cases that meet clinical diagnostic criteria.21
Diagnostic criteria for CHARGE syndrome.
| Classification |
|---|
| Typical: 3 major criteria or 2 major and 2 minor |
| Partial: 2 major and 1 minor criteria |
| Atypical: 2 major only or 1 major and 2 minor criteria |
| Major criteria |
| Coloboma (ocular) |
| Choanal atresia or stenosis |
| Hypoplasia or aplasia of semicircular canals |
| Minor criteria |
| Rhombencephalic dysfunction (brainstem and cranial nerve abnormalities) |
| Hypothalamic-pituitary dysfunction |
| Inner and/or outer ear malformation |
| Malformation of mediastinal organs (esophagus, heart) |
| Intellectual disability |
The most relevant differential diagnoses are DiGeorge syndrome, VACTERL association, Kabuki syndrome, teratogen-related embryopathies and oculo-auriculo-vertebral spectrum21 (Table 3).
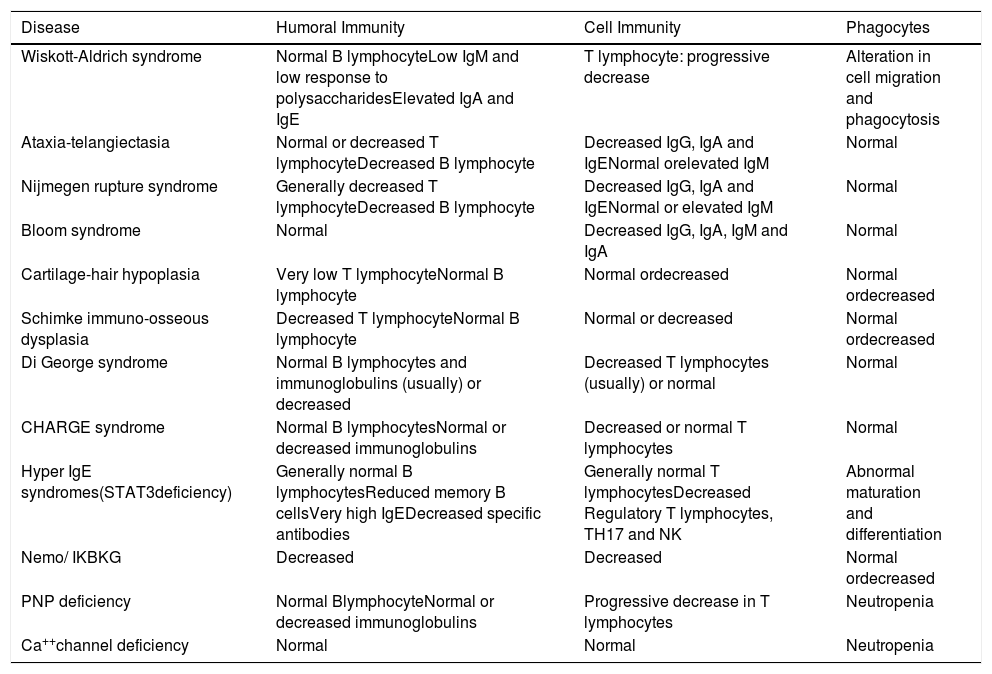
Immune defects in the main diseases classified as combined defects associated with syndromic characteristics.
| Disease | Humoral Immunity | Cell Immunity | Phagocytes |
|---|---|---|---|
| Wiskott-Aldrich syndrome | Normal B lymphocyteLow IgM and low response to polysaccharidesElevated IgA and IgE | T lymphocyte: progressive decrease | Alteration in cell migration and phagocytosis |
| Ataxia-telangiectasia | Normal or decreased T lymphocyteDecreased B lymphocyte | Decreased IgG, IgA and IgENormal orelevated IgM | Normal |
| Nijmegen rupture syndrome | Generally decreased T lymphocyteDecreased B lymphocyte | Decreased IgG, IgA and IgENormal or elevated IgM | Normal |
| Bloom syndrome | Normal | Decreased IgG, IgA, IgM and IgA | Normal |
| Cartilage-hair hypoplasia | Very low T lymphocyteNormal B lymphocyte | Normal ordecreased | Normal ordecreased |
| Schimke immuno-osseous dysplasia | Decreased T lymphocyteNormal B lymphocyte | Normal or decreased | Normal ordecreased |
| Di George syndrome | Normal B lymphocytes and immunoglobulins (usually) or decreased | Decreased T lymphocytes (usually) or normal | Normal |
| CHARGE syndrome | Normal B lymphocytesNormal or decreased immunoglobulins | Decreased or normal T lymphocytes | Normal |
| Hyper IgE syndromes(STAT3deficiency) | Generally normal B lymphocytesReduced memory B cellsVery high IgEDecreased specific antibodies | Generally normal T lymphocytesDecreased Regulatory T lymphocytes, TH17 and NK | Abnormal maturation and differentiation |
| Nemo/ IKBKG | Decreased | Decreased | Normal ordecreased |
| PNP deficiency | Normal BlymphocyteNormal or decreased immunoglobulins | Progressive decrease in T lymphocytes | Neutropenia |
| Ca++channel deficiency | Normal | Normal | Neutropenia |
The management of this disease is a challenge, as it involves continuous and multidisciplinary monitoring, since each condition that comprises the syndrome requires specific treatment.20,21,23
Hyper IgE syndrome (HIES)HIES is characterized by the triad eczema, recurrent infections and elevated serum IgE levels (> 5000 IU/mL).24,25 Several disorders have been successively described as an underlying cause of Hyper IgE syndrome: the autosomal dominant STAT3 deficiency and autosomal recessive deficiencies of DOCK8, PGM3 and ZNF431, in which atopy and elevated serum IgE levels occur in the context of manifestations not seen in patients with a STAT 3 mutation.25
a) STAT3 deficiencyPreviously called Job's syndrome, it is a disease with a heterozygous mutation that generates the loss of STAT3 function (OMIM 147060). Approximately 90 different mutations in STAT3 have been reported and most of them are missense mutations.25 STAT3 is widely expressed and mediates several pathways involved in wound healing, host defense and vascular remodeling, consistent with its multisystemic clinical phenotype. Multiple cytokines transduce the signal using STAT3, including IL-6, IL-10, IL-11, IL-17, IL-21, IL-22, IL-23, leukemia inhibiting factor, oncostatin M and cardiotrophin-1.21 The STAT3 loss-of-function mutations result in the failure to differentiate naive lymphocytes into Th17, which leads to Th2polarization.26
The first signs of HIES with loss of STAT3 function are atopic dermatitis of neonatal onset and increased susceptibility to infections by Staphylococcus aureus and Candida albicans. There are “cold” abscesses and lung abscesses caused by S. aureus or, more rarely, Streptococcus pneumoniae. Lung infections can also be caused by Aspergillus and Pseudomonas aeruginosa. Fungal infections, although severe, are usually limited to mucocutaneous surfaces and the lungs, such as chronic mucocutaneous candidiasis.20 Facial features become more evident during adolescence, with more prominent forehead and lower jaw, deep eyes, wide nose and porous skin. High-arched palate, retention of primary teeth and craniosynostosis are also common.21
This syndrome also comprises osteoarticular abnormalities, which include hyperextensible joints, scoliosis, small trauma fractures, osteopenia and degeneration of the cervical spine. Scoliosis can be severe enough to warrant surgical correction. Unlike the poor wound healing seen after pulmonary surgery, orthopedic surgery is generally not complicated. The joints are typically hyperextensible, which may explain the significant arthritis seen at younger ages, when compared to the general population. Degenerative cervical spine disease in the fourth and fifth decades of life can cause neurological deficits and require surgical stabilization.20,21 These patients may also have vascular abnormalities, such as aneurysms, myocardial infarction, subarachnoid hemorrhage and intestinal hemorrhage. Asymptomatic central nervous system abnormalities are a common finding on MRI images of the brain in these patients, such as focal enhancement of white matter.21
Approximately 7 to 9% of patients also develop malignancies, especially several types of lymphoma, including Burkitt, non-Hodgkin, histiocytic, peripheral T-cell lymphoma and other B-cell lymphomas.20
The most consistent laboratory finding is the high level of serum IgE. The peak is usually > 2,000 IU/mL, but the level tends to decrease or even return to normal with increasing age and does not correlate with disease severity. The CBC is usually normal, but relative neutropenia may be present. The white blood cell count often does not increase in response to infection. The presence of eosinophilia is also common. Although serum IgG and IgM are normal and serum IgA is normal or low, the response of specific antibodies to encapsulated microorganisms may be impaired. Lymphocyte phenotyping often reveals very low memory T and B cells and very low IL-17-producing T cells.20
The main goal of therapy for Hyper IgE syndrome due to STAT3 deficiency involves the prevention and treatment of infections. Prophylactic antibiotics directed at S. aureus are useful to decrease the frequency of pyogenic pneumonia and thus prevent the formation of parenchymal lung disease, as well as skin involvement. Anti-Aspergillus prophylaxis should also be considered for any patient with pneumatocele, due to the increased risk of developing aspergillomas. Human immunoglobulin replacement therapy can decrease the incidence of sinopulmonary infections.26 Despite the high morbidity of this disease, approximately 80% of the patients live up to 50 years of age.25
b) DOCK8 deficiencyDOCK8 deficiency (OMIM 243700) is part of the scope of Hyper IgE syndromes. It is caused by autosomal recessive loss-of-function mutations of this gene involving, in most cases, large gene deletions. The DOCK8 protein regulates the cytoskeleton rearrangement necessary for cell migration and the formation of immune synapses, reflecting the innate and adaptive immunity.24 These mutations result in combined immunodeficiency, autoimmunity and atopy.24,25
Similarly to patients with a STAT3 mutation, patients have atopic dermatitis, skin infections by S. aureus, pneumonia, elevated serum IgE levels and eosinophilia. Normally, there are more allergic manifestations in DOCK8 than in STAT3 deficiency, which are even more severe, with food allergies, asthma and eosinophilic esophagitis.25,26 There is a susceptibility to cutaneous viral infections, such as human papillomavirus (HPV), resulting in disseminated and recalcitrant warts, disseminated contagious molluscum, lesions by the Herpes Simplex virus and the Varicella zoster virus. Most patients also have a history of recurrent sinopulmonary infections, including Pneumocystis jiroveci pneumonia. Complications such as the formation of bronchiectasis occur in more than one-third of patients. Malignancy is a fundamental characteristic of DOCK8 deficiency, appearing at a younger age and with aggressive tendencies. Malignant diseases result from immune dysregulation and are related to viruses.21 Abnormalities in skeletal, dental and connective tissue are absent. Neurological manifestations of infectious and non-infectious causes (vasculitis, vascular aneurysms and cerebral infarctions) are frequent.24,25
Elevated IgE levels (mean 2,000 IU/mL), eosinophilia and T-cell and NK lymphopenia are found in most patients. B-cell alterations occur less frequently, although IgM levels are typically decreased and decline with age. Differentiation of Th17 cells is also impaired.25,26 An image scan of the brain vessels to detect areas of stenosis is essential to prevent stroke.26
To make the diagnosis, in addition to the clinical picture and laboratory tests, the analysis of flow cytometry for the expression of the DOCK8 protein is a reliable diagnostic test, as most patients have mutations that interfere with the normal protein expression. Analysis of genetic material by Sanger sequencing can be difficult due to the prevalence of large deletions in the gene.24
Hematopoietic stem cell transplantation has shown to be the only curative option for DOCK8 deficiency and is recommended in the early stages of the disease. While the patient awaits definitive treatment with the transplantation, significant complications related to infections and malignancy must be quickly treated. Antibacterial and antiviral prophylaxis and human immunoglobulin replacement therapy are recommended.26
c) PMG3 deficiencyFirst described in 2014, phosphoglucomutase 3 (PGM3)deficiency is a congenital glycosylation disorder.25 Generalized clinical manifestations are believed to be due to the role of glycosylation in normal cell functions, being particularly necessary for the normal function of most immune receptors, immunoglobulins, complement proteins and cytokines.26 PMG3 deficiency has an autosomal recessive inheritance pattern, with mutations of different types, thus resulting in immunological and clinical defects of different severities.25,27
The clinical phenotype of patients is quite variable, with two main poles: severe combined immunodeficiency and hyper IgE syndrome.26 Patients with these two phenotypes have common clinical features, including severe infections (recurrent respiratory infections, gastrointestinal tract infections and, less commonly, abscesses and candidiasis), glomerulonephritis (possibly due to autoimmunity) and atopic disease (eczema, asthma, multiple allergies). The most common immunological alterations are neutropenia, lymphopenia and eosinophilia with varying severity. Patients with the hyper IgE phenotype have increased serum IgE levels and a T-cell disorder with a Th2 bias similar to that of patients with DOCK8 deficiency.25 They also have various severe non-immunological abnormalities, including dysmorphic characteristics, skeletal dysplasia, scoliosis, brachydactyly and delayed neuropsychomotor development.25,27 The definitive diagnosis is molecular, and the treatment is directed at infections’control with prophylactic antibiotic therapy and human immunoglobulin infusion.27
d) ZNF431 deficiencyThe ZNF431 gene is located on chromosome 20q11.22. and encodes a transcription factor that acts as a positive regulator of STAT3 expression. Autosomal recessive homozygous mutations in ZNF431 lead to insufficient levels of STAT3, generating a phenotype similar to autosomal dominant hyper IgE syndrome.28 This mutation creates a defect in the differentiation of naive T cells into Th17 cells (and therefore in the production of IL17), resulting in decreased Th17, excess of Th2 response, and decreased memory B cells.24,28 Although these patients have high levels of IgE, to date, the ZNF431 gene has not been associated with atopy traits, such as asthma, rhinitis, atopic dermatitis and allergic sensitization.28 There is no facial dysmorphism, dental and bone alterations or pneumatoceles.24
Anhidrotic ectodermal dysplasia with immunodeficiencyThe NF-κB transcription factor comprises homo- or heterodimer protein complexes consisting of NF-κB1/p50 (p105precursor), NF-κB2/p52 (p100precursor), RelA/p65, RelB ou c-Rel,and it translocates to the nucleus after activation of the canonical or non-canonical NF-κB pathway.
Anhydrotic ectodermal dysplasia with immunodeficiency (EDA-ID) or NEMO / IKBKG deficiency is an autosomal dominant or X-linked recessive genetic disease.1 The occurrence of the X-linked recessive type of EDA-ID is estimated to be 1 in 250,000 individuals (NIH, 2020).
Affected individuals have severe recurrent infections in early childhood, with eczema, hypotrichosis, hypodontia or small, sharp teeth. Most children have hypohidrosis, because they have fewer sweat glands or glands with impaired function. Some patients with anhidrosis may experience a dangerous increase in body temperature (hyperthermia), especially in hot climates and during exercise. Children with EDA-ID usually have pneumonia, otitis media, sinusitis, lymphadenitis, skin and bone alterations. Approximately a quarter of individuals with EDA-ID have typical inflammatory alterations, such as inflammatory bowel disease or rheumatoid arthritis.29
The laboratory findings described in most patients with NEMO deficiency are: reduced levels of IgG (especially IgG2), aberrant levels of IgM and IgA, deficient response of specific polysaccharide antibodies, low number of Class-switched memory B cells and impaired IL-10 production after activation with tumor necrosis factor α (TNF-α). Patients with NEMO deficiency with severely compromised T-cell function are at high risk for life-threatening infections and may benefit from early BMT, while patients with NEMO deficiency without combined immunodeficiency may benefit from conservative therapy.30
OthersPNP deficiencyPurine nucleoside phosphorylase (PNP) deficiency (OMIM 613179) is a rare, autosomal recessive disorder.31 The PNP gene is located on chromosome 14q13.1 and the PNP enzyme is important for the degradation and recovery of purine, an extremely relevant process for energy production and DNA formation.32,33 This enzyme is expressed in practically all cells, but with more intensity in lymphoid cells.31,33 Its deficiency results in the gradual intracellular accumulation of substances with a toxic effect, mainly in thymocytes, resulting in the dysfunctional development of these cells and of cell immunity components, in addition to apoptosis of neuronal cells.32
The classic clinical picture is a late-onset immunodeficiency,31 associated with neurological abnormalities in two-thirds of the cases and autoimmune diseases in one-third of them. The immunodeficiency is the severe combined type (SCID) with a widely variable age of onset and disease course, with many patients being diagnosed only after the second year of life. There is a progressive decline in the number of T lymphocytes, but B and NK cells are usually spared (SCID T-B+NK+).31,34 Neutropenia occurs, frequently related to autoimmunity or infections.33 Another common laboratory finding is the presence of hypouricemia, which may represent a screening test.35
Patients usually have multiple infectious manifestations, such as viral, bacterial and fungal infections. In the first decade of life, they can be caused by opportunistic pathogens, including Aspergillus fumigatus, Mycobacterium and the John Cunningham virus. Autoimmune manifestations occur, such as autoimmune hemolytic anemia, idiopathic thrombocytopenic purpura, autoimmune thyroiditis, autoimmune neutropenia, autoimmune cholangitis, pericarditis and arthritis. Neurological abnormalities range from mild delayed neuropsychomotor development to mental retardation. Ataxia, imbalance, spastic paresis, hyper or hypotonia can also be seen. Sensorineural hearing loss has been documented in some cases.31–33
Early patient identification, as well as rapid treatment initiation will be crucial in preventing irreversible toxicity to many organs, including the thymus, brain and lungs.33
Hematopoietic stem cell transplantation is the only curative treatment for these patients.32 However, there are a few reports of transplantation in patients with PNP deficiency, possibly reflecting the hesitation to transplant these patients who are usually diagnosed after early childhood and suffer from multiple organ damage, including neurological abnormalities. Currently, there are no gene therapy trials for patients with PNP deficiency.33
Calcium channel defectsImmunodeficiency-10 (IMD10) or STIM1 deficiency (OMIN 612783); and immunodeficiency-9 (IMD9) or Oral1 deficiency (OMIM: 612782) are extremely rare diseases (approximately 6 described patients), of which inheritance is autosomal recessive. In IMD 9, infections are due to defective T-cell activation.1 IMD10 is characterized by the onset of recurrent infections in childhood due to the deficient T- and NK-cell function, resulting in potentially fatal infections by viral, bacterial and fungal pathogens. Also, in IMD10, hypotonia, hypohidrosis or hypoplasia of the tooth enamel consistent with imperfect amelogenesis can be observed.36 The pathogenesis of the disease is related to calcium channel defects.36,37
Conflicts of interestThe authors declare no conflicts of interest.
Study conducted at Centro Universitário Saúde ABC, Faculdade de Medicina, Serviço de Referência em Doenças Raras, Santo André, São Paulo, SP, Brazil.





