


To rescue medical genetics concepts that are necessary to understand the advances in the genetic-molecular characterization of primary immunodeficiencies, to help in the understanding and adequate interpretation of their results.
Source of dataNon-systematic literature review, searching for articles since 2000 on PubMed using the terms “genetic evaluation” OR “whole exome sequence” or “whole genome sequence” OR “next generation sequence” AND “immunologic deficiency syndromes” OR “Immune deficiency disease” OR “immune deficiency” NOT HIV.
Summary of the dataKnowledge of medical genetics is essential for the understanding of the principles of heredity and disease inheritance patterns, types of genetic variants, types of genetic sequencing and interpretation of their results. The clinical and immunophenotypic evaluation of each patient is essential for the correlation with the genetic variants observed in the genetic study of patients with primary immunodeficiencies. The discussion of the benefits and limitations of genetic tests should always guide the performance of genetic tests.
ConclusionsThere are many evident benefits of genetic analysis, such as the definitive diagnosis of the disease, family genetic counseling, and the possibility of a more adequate and accurate management. Cost, access and interpretation of genetic test results are limitations that need continuous improvement. The understanding of the benefits and limits of the several genetic assessment methodologies related to primary immunodeficiencies is essential to obtain more effective results from the sequencing.
Primary immune deficiencies (PID) are diseases caused by monogenic germ line mutations in genes related to the immune system, which result in loss of expression and loss of function (LOF), amorphic or hypomorphic, or gain of function (GOF, hypermorphic) of the encoded protein. PIDs, also recently classified as Inborn Errors of Immunity (IEI), manifest as being more susceptible to infectious diseases, autoimmunity, auto inflammatory diseases, allergies, and/or malignancies.1,2
PIDs were considered rare diseases, with an estimated prevalence of 1 in 10,000 to 1 in 50,000 live births. However, recent advances in platforms for genetic studies and their interface with immunological knowledge have allowed a greater understanding of immuno-genetics, which culminated in the discovery of the causing genes of phenotypically-known PIDs and many other new PIDs.3,4 In this sense, more recent studies have shown that PIDs are much more frequent than previously estimated and that around 1% of the population has some form of PID when all types and variations are considered.5,6
This great advance in the knowledge of PIDs occurred in parallel to the Human Genome Project, with the development of the next-generation DNA sequencing technologies (NGS, Next-Generation Sequencing), which allowed the study of a large number of genes at the same time, including the study of the whole exome or even the whole human genome. These genetic sequencing platforms are responsible for the generation of a massive database; then, the development of computational analysis platforms took place, so that all these generated data could be analyzed in an adequate and faster way, which has been called bioinformatics or biocomputing, a specialty that combines the knowledge of biology and informatics to a high standard. In other words, these new platforms have led to a huge leap forward in the identification and diagnosis of previously inconclusive immune system defects.3,7
The International Union of Immunological Societies (IUIS), with the objective of monitoring, following and cataloging the recent discoveries published in the medical literature, has gathered a highly-qualified medical team, which has organized biannual publications aiming at the dissemination of knowledge regarding all advances, new diseases and genes involved in the field of PIDs. The most recent publication, from the beginning of the year 2020,showsan increase in the number of these immunological diseases to 406, with 430 known genetic defects identified as causing these conditions.1
The objective of this review article is to rescue concepts of medical genetics that are necessary to understand the advances in the genetic-molecular characterization of primary immunodeficiencies, aiming to help the understanding of the currently available types of genetic evaluations and the interpretation of their results, as well as the importance of medical knowledge for the achievement of clinical correlation with these findings.
Recalling basic concepts of medical geneticsThe basic concepts of genetics are essential for the broad understanding of the PID-related inheritance and also its pathophysiology. Initially, let us remember that the human being has 23 pairs of chromosomes, of which 1 pair determines the gender and is called the sex chromosome, and another 22 pairs of chromosomes called autosomal, which contain the genes, responsible for our entire structural and functional framework. The genetic material is transmitted to each individual in 2 parts, a maternal and a paternal one, known as the maternal and paternal alleles within each gene.8,9
Each gene consists of regions known as exons, responsible for gene transcription, and introns, regions that have structural and DNA support functions, which alternate throughout the gene. During the transcription process, only the exon content is used in the creation of the messenger RNA through a structural rearrangement of the DNA. The genetic material present in the exons and introns is formed by nitrogenous bases known as adenine, thymine, guanine, and cytosine, which are repeated millions of times in our DNA. Each set of sequences of 3 nitrogenous bases located in the exons is called a codon and leads to the placing of an amino acid in the protein formed at the end by the gene. This sequence of base pairs is conserved in most individuals; however, some regions may show some alterations without harming the final protein product, known as genetic variants. These variations are found at different proportions in the population and can be accessed in genetic databases of the human genome.8,9
However, some errors in this sequence of base pairs can lead to a change in an amino acid that is essential for some protein function, leading to loss of its function (LoF) or excessive activity of the protein (GoF). Other times, these changes can lead to the formation of a stop codon, responsible for interrupting transcription before the total formation of mRNA, leading to an early stop of the protein formation and, consequently, of its stability and function, making it many times undetectable. Other errors in this sequence of nitrogenous bases can occur, such as insertions or small deletions, which change the entire codon sequence starting from an alteration and consequently the entire structure and function of the formed protein. In these situations, these changes in nitrogenous bases are called pathogenic variants or mutations.9
Another concept of the utmost importance in medical genetics is that of the pattern of inheritance. These patterns of inheritance are an important part of the family history of patients with suspected PID and are essential to the performance of genetic counseling. The autosomal recessive (AR) pattern is one where a given phenotype is expressed when the two alleles of a given gene are altered. This is the pattern observed in most PIDs and the majority of them are related to the loss of function of that gene. In the AR pattern, the pathogenic genetic variants can be similar in the 2 alleles and are thus described as being homozygous, or each allele can have a distinct genetic variant that leads to its loss of function, with this presentation being described as compound heterozygous.1,2
In the autosomal dominant (AD) pattern, a certain clinical phenotype is expressed by an alteration in only 1 of the alleles that make up a given gene. It can occur as a familial trait, that is, one of the parents has a similar alteration, or by de novo mutations, which occur in the gametes and are transmitted directly to the descendants, even if the parents do not have the genetic variant. This pattern occurs in several PIDs, such as Hyper IgE Syndrome, CTLA4 haplo insufficiency, and APDS, among others. Sex-linked inheritance or X-linked inheritance is related to genes present on the non-homologous part of the sex chromosomes, that is, genes present on the X chromosome that have no counterparts on the Y chromosome. Since male individuals have only one chromosome X and this non-homologous region has no alleles, they are called hemizygotic. Some of the classic immunodeficiencies have this pattern of inheritance, such as X-linked Agammaglobulinemia (XLA), Wiskott-Aldrich Syndrome, Severe combined immunodeficiency (SCID) due to gamma chain deficiency, X-linked DGC, and IPEX syndrome, among others.1,2
Understanding the basic notions of genetics is essential to understanding the family history of patients with PID, as well as for the proper interpretation of the genetic reports that are normally requested and how genetic counseling should be carried out in each case.
Genetic variantsGenetic sequencing analyzes the sequence of nitrogenous base pairs present in the assessed DNA, which is compared with the databases assembled from the Human Genome Project, most of which are currently freely available. Genetic variation is the difference in DNA sequences between the individuals in a population and occurs in germ cells, that is, sperm and eggs, and also in somatic cells. Only the variation that appears in the germ cells can be inherited from one individual to another and, therefore, affect the population dynamics and, ultimately, evolution.10
Mutations are the original source of genetic variation. A mutation is a permanent change in a DNA sequence. De novo (new) mutations occur when there is an error during DNA replication that is not corrected by the repair enzymes and, therefore, this error is copied and fixed in the DNA. Mutations can be neutral for the body; in some situations, they can be beneficial; and at other times, harmful, and can lead to clinically evident diseases, that is, pathogenic ones. Recombination is another important source of genetic variation. Each individual has a mixture of genetic material from their parents that occurs during recombination, when the homologous strands of DNA align and cross over and can create new combinations of variants in the offspring germ cells.10
The term genetic variant is used to define specific regions of the genome that differ from the genome present in the databases. Variants can be named according to the type of exchange and the consequent alteration caused by that exchange.
- •
Silent variant: a single exchange of a nitrogenous base for another occurs, which alter the codon, but, does not change the coded amino acid, that is, the protein remains unchanged. For that to occur, it is important to remember that an amino acids can be encoded by different sequences of nitrogenous bases. Overall, these variants do not cause problems for the patients, that is, they are considered benign.
- •
Missense variant: there is a single exchange of a nitrogenous base for another that alters the encoding of the amino acid in that position. The protein undergoes an alteration in its linear structure that may or may not impact its stability and function. Functional studies are necessary to verify whether that protein underwent functional alteration or not.
- •
Nonsense variant: there is a single exchange of a nitrogenous base and it changes the codon of the aminoacid to a translation stop codon, known as a stop codon. From the point of exchange no amino acids are added, and in most cases the formed protein is unstable and/or dysfunctional.
- •
Frameshift variant: in this group of variants there are insertions or deletions of nitrogenous bases that change the entire underlying sequence of codons, that is, they change the amino acids that will be added from that point on, generating a completely different protein. They are also called INDEL (insertion and/or deletion). Most of them are unstable and/or dysfunctional.
- •
Splicing variant: these are variants that promote changes in the genetic rearrangement between exons and introns for the creation of mRNA. Overall, they promote the formation of altered messenger RNA and, consequently, altered proteins. Variants at bases 1 and 2 in the transition region between exons and introns are the most associated with this type of alteration. Most of these altered proteins are unstable and/or dysfunctional.
Initially, for a universal understanding, the variants detected in a genetic sequencing must be reported following the international nomenclature rules indicated by the Human Genome Variation Society (HGVS). The nomenclature is also important in cases where the variant needs to be screened in the patient's family members, ensuring that the same variant that was previously detected is being evaluated and thus avoiding a false negative result.11
In studies of human genetics, genetic variants are further classified according to the effect induced by this modification of the genome regarding the function of that gene and, consequently, the possibility of causing or not causing diseases in individuals. Therefore, the variants can receive the following nomenclatures: pathogenic, probably pathogenic, variant of uncertain significance (VUS), probably benign, and benign. Pathogenic variants are those in which the presence of a studied gene function alteration and the patient's clinical disease have already been studied and confirmed in a laboratory and/or clinically. The probably pathogenic variants are those found in genes known to cause a certain disease that is under suspicion, and the type of variant is highly indicative of loss of function of the gene, such as the INDEL type, stop codon, and splicing variants. On the other hand, benign variants are those studied and confirmed clinically and/or in a laboratory, in which there was no loss of gene function due to the variation. Also considered to be benign are those variants that show a high frequency in the population, generally higher than 1%, which would not occur if they were pathogenic. Probably benign variants are those that are not expected to have any pathogenicity (e.g. silent and/or intronic variants) but do not meet all the criteria to be classified as benign.
The group of variants that show the greatest difficulty are those called variants of uncertain significance. They are variants whose available scientific evidence does not allow to conclude whether they are benign or pathogenic variants. They are usually rare variants, absent or found at a low frequency in population genetic databases, without previous reports and whose evaluations carried out by protein function modification computer programs show conflicting results (in silico). These are variants that would require laboratory research to verify the effect of the mutation on the final result and on the protein function to be formed, which we cannot do in clinical practice because it requires highly specialized laboratories.11
Genetic sequencing techniquesGenetic sequencing is the study of the sequence of nitrogen base pairs that form our DNA. Regarding the investigation of diseases, we look for changes in this sequence that interfere with the formation of messenger RNA, and therefore in the final product of genes, the proteins that perform several functions in our bodies. When requesting a genetic evaluation, it is important to know what we are looking for, since there are several available genetic tests that have different methodologies, different objectives and a different number of genes that can be studied. Therefore, we have chosen to carry out a small review aimed at the ones that work with the clinical part of the currently available genetic testing options to help diagnose the patients, available for patients with PID and also for other diseases.12
Sanger techniqueThe Sanger technique was the first to be described and is still widely used in several basic genetic studies, but it has lost a lot of space in clinical genetics in recent years. The genetic sequencing used in this technique is a manual one, carried out in small pieces, mostly exon by exon of each gene. Due to these characteristics, it has a high cost per studied gene, cannot be automated, and is very laborious. In investigations of PIDs, where several genes can lead to similar symptoms, it becomes a procedure with high cost and longer performance time. It is still a very useful technique and performed for specific situations, such as the study of genes that have pseudo genes or for confirmation in situations that conflict with the use of NGS techniques. It can also be a useful technique when diseases caused by just one gene are suspected.13
Next-Generation sequencing (NGS)The NGS was conceived based on the need to automate the process of genetic sequencing, thus allowing a large volume of genes to be analyzed at the same time. In this technique, many standard structures that bind to pieces of DNA, known as primers, are used, amplifying them and allowing their analysis. In this process, the data from the nitrogenous bases are converted into binary sequences and the result of the process is analyzed computationally with a large volume of data. Therefore, this process requires investment in bioinformatics, in addition to the laboratory. The NGS allows us different levels of study according to the interest of ongoing genetic research and has been extremely useful in assessing primary immunodeficiencies.13
Targeted sequencing or genetic panelsTargeted sequencing, also known as genetic panels, is now widely used by most clinical genetic laboratories. In this type of sequencing, one can study groups of genes that are associated with the patients’ clinical manifestations, used for almost all areas of medicine. Because it has a limited number of genes, the panel study is less costly, due to the reduction in the used materials and a smaller bioinformatics analysis.14 Another advantage of this technique is the possibility of carrying out a quantitative analysis of the genes’ amplification, which allows us to confirm the presence of large deletions/common insertions as the genetic cause of several IEI (DOCK8, LRBA, for instance), an analysis known as copy number variation (CNV).15,16 It is also important to remember that most panels only study the exons of these genes. The genetic panels for PIDs can miss genes that are not contained in them, as well as genes located in non-coding regions, that is, intronic ones.13 For doctors in clinical practice, it is important to know that the genetics laboratory sets up a different panel, and sometimes that gene (or genes) that one would need to investigate may not be present on that panel. Before making a request, it is necessary to know the panel of the institution that will carry out the test and, particularly, whether it contemplates the genes one wants to analyze.12,13
Whole exome sequencing (WES)WES sequences the exons of all the genes in our DNA, which produces a huge amount of data for bioinformatics analysis. In a simplified manner, a great advantage of this test would be the possibility to evaluate all genes at the same time. However, many considerations must be made in relation to the cost of the test, much higher than the carrying out of a given panel; the large number of variants that can be found related to other processes in our body that must be evaluated based on the result; and, furthermore, a reduction in the capacity to assess the CNVs, which were previously described.12
WES is currently available in most clinical genetic laboratories, but from a practical and economic point of view it has been mainly used in research. Some clinical laboratories run WES only; however, they perform data analysis according to the medical request, reducing the cost and time for data analysis. The growing improvement in the bioinformatics analysis process, the presence of an ever growing international database, fed by the sequencing of different populations around the world, and a decrease in the prices of the materials used might lead us in the coming years to a cost very close to that of genetic panels, making it more accessible from a clinical point of view.13
Whole genome sequencing (WGS)WGS is the sequencing of all DNA, including the exons and introns of all genes. It is a widely used technique to characterize a species and its variants. It has been extensively used during the COVID-19 pandemic for the characterization of the viruses in different regions, helping to understand changes that have been occurring with the SARS-CoV-2 during the pandemic. For the study of IEI, it has been used for the investigation of new diseases. Its disadvantage is its high cost, in addition to the need for a wide bioinformatics apparatus to perform data analysis, remembering that we will have billions of letters to be analyzed for each individual, in an attempt to find an error in this sequence that is not just a variation from the normal.13
Hybridization and micro-arrayCopy number variation (CNV) constitutes the number of copies of a given gene that varies from one individual to another. The Human Genome Project demonstrated that the genome experiences gains and losses of genetic material, which can be associated with pathologies in human beings. In practical terms, these variations may comprise large deletions, which would reduce the number of copies that are amplified in a sequencing, as they would amplify only one allele.17 Despite the development of bioinformatics tools for the detection of these CNVs in NGS, the gold standard for genetic diagnosis continues to be genomic hybridization by microarray. In the comparative genomic hybridization (CGH), the hybridization of the patient's DNA with a microarray of DNA oligonucleotide probes is compared to the competitive DNA hybridization with the probes in the same assay, and the probes are generally designed specifically for the detection of CNVs.18
Genetics in inborn errors of immunityAs stated earlier, advances in the field of genetic sequencing technologies were fundamental for a greater understanding of PIDs. The scientific progress generated by all these studies has been accompanied by significant benefits in the clinical area, since when one reaches the molecular diagnosis, one can better understand the pathophysiology of the disease and the correlation with the patient’s clinical condition. In many cases, this understanding allows us to choose specific treatment options for each condition, inside the concept of Precision Medicine. However, from the working bench to the bedside, or rather, the medical office, there are challenges to be overcome.
Currently, more than 430 genes have already been described as monogenic causes of primary immune deficiencies, and that number is expected to grow even more in the coming years. The interface between immunology and genetics has become extremely productive, and the need for knowledge of genetic sequencing methodologies by those medical professionals who treat patients with PID is clear, because only then we will understand the great difference in tests in genetics, and also the great difference regarding the costs of these sequencing methodologies in different laboratories. Based on this interface, professionals treating a patient with suspected PID should perform a clinical and laboratory evaluation according to their clinical suspicions, and if according to their initial findings the genetic investigation can provide diagnostic confirmation or exclusion, or provide the possibility of specific treatments or even the access to those treatments, we must think about sequencing for the patient.13
To highlight the importance of genetic sequencing in the field of PIDs, a recent study found that in 60 (55%) out of 110 families there was a change in the diagnosis of their initial PID based on clinical immune phenol typing data after the molecular diagnosis was performed by WES. The same study also found that in 25% of cases, that is, in 26 out of these 110 families, there was a substantial change in the management of patients with PID, with at least 14 patients undergoing hematopoietic cell transplantation after the genetic investigation findings with WES.19 In addition to the indication of bone marrow transplantation (BMT) for a large number of PIDs, the molecular diagnosis can direct a more precise treatment according to the affected immunological pathway, such as the use of abatacept in CTLA4 and LRBA deficiencies,20,21 the use of Janus-Associated Kinase/STAT inhibitors in conditions of STAT1 and STAT3function gains,22,23 the use of AKT-mTOR pathway inhibitors in patients with PIK3 pathway changes with increased activity,24 the use of IL1 antagonists in patients with some autoinflammatory diseases,25 and interferon alfa for defects of the TLR3 pathway,26 among others.
On the other hand, the same previously mentioned study found no genetic changes that could confirm the molecular diagnosis of PID in 60% of the 278 participating families.26 The presence of gene regions without adequate coverage, including regulatory regions and polyA, low-grade mosaicisms, small CNV and INDEL, intronic mutations and the presence of pseudogenes may be associated with not finding variants in these cases.26,27 As an example, the currently most commonly used WES platforms have less than 100% coverage in94 genes in the IUIS list, less than 99% in at least 26 genes, and five with less than 90% coverage (IKBKB, NCF1, TACI, UNC93B1, and TBX1);in other words, part of the genes are not sequenced and analyzed and, therefore, genetic variants in these regions are not identified.27
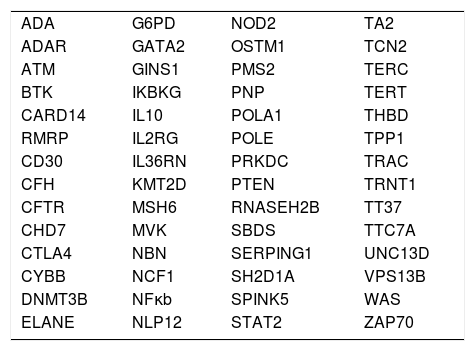
Table 1 shows a series of genes present in the IUIS list that have intronic pathogenic or probably pathogenic mutations already described in the ClinVar database. Table 2 shows the genes present in the IUIS list that have at least one pseudo gene, which can interfere with the appropriate interpretation of their sequence.
Genes related to primary immunodeficiencies present in the list of theInternational Union of Immunological Societies(IUIS) with pathogenic or probably pathogenic variants located in nonexonic regions listed in ClinVar.
| ADA | G6PD | NOD2 | TA2 |
| ADAR | GATA2 | OSTM1 | TCN2 |
| ATM | GINS1 | PMS2 | TERC |
| BTK | IKBKG | PNP | TERT |
| CARD14 | IL10 | POLA1 | THBD |
| RMRP | IL2RG | POLE | TPP1 |
| CD30 | IL36RN | PRKDC | TRAC |
| CFH | KMT2D | PTEN | TRNT1 |
| CFTR | MSH6 | RNASEH2B | TT37 |
| CHD7 | MVK | SBDS | TTC7A |
| CTLA4 | NBN | SERPING1 | UNC13D |
| CYBB | NCF1 | SH2D1A | VPS13B |
| DNMT3B | NFκb | SPINK5 | WAS |
| ELANE | NLP12 | STAT2 | ZAP70 |
Genes related to primary immunodeficiencies present in the list of the International Union of Immunological Societies(IUIS) that have at least one pseudogene.
| IUIS genes with 1 pseudogene | C19orf40, CD46, CDCA7, CSF2Rb, DCLRE1C, FPR1, HAX1, IKBKG, ITCH, MAGT1, MSN, MTHFD1, NBAS, NCSTN, NHP2, NOP10, PIK3CD, PNP, PTEN, RANBP2, RLTBP2, RLTPR, RNASEH2C, RTEL1, TCF3, TMC6, ZBTB24 |
|---|---|
| IUIS genes with more than 1 pseudogene | ACTB, AK2, CFTR, IGLL1, NCF1, PMS2, RAC2, RNF168, RPSA, SBDS, TRNT1, UNG, XIAP |
Due to the number of citations in this review, it is important to mention that IUIS brings a review of all new genes associated with PIDs every two years, as well as the phenotypic expansion for genetic defects that have already been characterized, and is an essential source for routine consultation with patients and when requesting genetic evaluations.1
ConclusionsGenetic assessment in the field of primary immune deficiencies has brought unprecedented advances in patient knowledge and management. In the clinical practice, the benefits are evident, such as definitive diagnosis, family genetic counseling and the possibility of a more adequate and accurate approach, improving the quality of life and reducing risks of death and sequelae for patients with PID. On the other hand, there are several barriers that need to be overcome, such as cost, access to tests and interpretation of genetic results. It is necessary that physicians who treat patients with PID be able to understand more and more the benefits and limits of the different methods of genetic evaluation and can direct the investigation in the most appropriate way according to the clinical and immune phenotypic evaluation of each patient, thus increasing the chance of more effective sequencing results.
FundingThe Jeffrey Modell Foundation CHILDREN program.
Conflicts of interestThe author declares no conflicts of interest.





