





The abnormalities of the genitourinary tract development are the leading cause of chronic kidney disease (CKD) in children. The diagnosis of this disease in Brazil is late and incomplete, which results in increased morbidity and mortality in this age group. Early diagnosis of this condition is the prerogative of generalist pediatricians, and the aim of this study was to review the clinical signs and symptoms associated with developmental abnormalities of the genitourinary tract.
Data sourcesBased on the description of a symbolic clinical case, the authors conducted a non‐systematic review of medical literature.
Data synthesisThe results suggest that the following data should be used as a warning for early diagnosis of affected children: a) combined urinary tract abnormalities (chromosomal abnormalities; sequence of malformations [VACTERLand Prune‐Belly]; and musculoskeletal, digestive tract, heart, and nervous system malformations); b) previous history (congenital anomalies of the kidney and urinary tract [CAKUT] in the family, low birth weight, and oligoamnios); c) clinical signs (polyuria/nocturia, urinary tract infection, systemic arterial hypertension, failure to thrive, weak urinary stream, difficulty to start urination, distended bladder, non‐monosymptomatic enuresis, urinary/urge incontinence, and bowel and bladder dysfunction); and d) pre‐ and postnatal ultrasonographic alterations (increased anteroposterior diameter of the renal pelvis, mainly in the third trimester of pregnancy; single kidney; hydronephrosis associated with other abnormalities; and hydronephrosis with parenchymal involvement in the post‐neonatal assessment).
ConclusionThe suggestions shown here can help the pediatrician to establish clinical hypotheses for the early diagnosis of developmental abnormalities of the genitourinary tract without resorting to expensive and invasive procedures.
As anormalidades do desenvolvimento do trato geniturinário são a principal causa de doença renal crônica (DRC) em crianças. O diagnóstico dessa doença no Brasil é formulado de maneira incompleta e tardia, o que resulta em aumento na morbimortalidade nessa faixa etária. O diagnóstico precoce dessa condição é prerrogativa dos pediatras generalistas e o objetivo deste trabalho foi revisar os sinais e sintomas clínicos associados às anormalidades do desenvolvimento do trato geniturinário.
Fontes dos dadosA partir da descrição de um caso clínico simbólico, fizemos uma revisão não sistemática da literatura médica.
Síntese dos dadosOs resultados sugerem que os seguintes dados devem ser usados como alerta para o diagnóstico precoce das crianças acometidas: a) anomalias do trato urinário compostas (anomalias cromossômicas, sequências de malformações – Vacterl e Prune‐Belly, malformações musculoesqueléticas, do trato digestivo, cardíacas e do sistema nervoso); b) antecedentes (anomalias congênitas do rim e trato urinário (CAKUT) na família, baixo peso ao nascer e oligoâmnio); c) sinais clínicos (polaciúria/noctúria, infecção urinária, hipertensão arterial sistêmica, baixo ganho de peso, jato urinário fraco, dificuldade para iniciar a micção, bexigoma, enurese não monossintomática, urge/incontinência urinária, disfunção do intestino e da bexiga) e d) alterações ultrassonográficas ante e pós‐natais (diâmetro anteroposterior da pélvis renal aumentado principalmente no terceiro trimestre da gestação, rim único, hidronefrose associada a outras anomalias e hidronefrose com comprometimento de parênquima na avaliação pós‐neonatal).
ConclusãoAs sugestões apresentadas podem ajudar o pediatra a estabelecer hipóteses clínicas para o diagnóstico precoce das anormalidades do desenvolvimento do trato geniturinário sem metodologias caras e invasivas.
H.O.R.S, 12 anos e três meses, nasceu a termo, sem intercorrências durante o parto. Recebeu alta médica em 72h na companhia da mãe com orientação de seguir acompanhamento com especialista, pois em ultrassonografia pré‐natal havia sido evidenciada hidronefrose bilateral com medida de diâmetro anteroposterior da pelve renal de aproximadamente 12mm. Porém, após o nascimento, os pais perderam o seguimento médico da criança com o nefrologista.
Compareciam às consultas com o pediatra irregularmente. Aos oito anos foi encaminhado para consultas com o hematologista e endocrinologista devido à baixa estatura e anemia não responsiva ao tratamento com ferro por via oral; esses especialistas não encontraram a causa para as alterações.
Apenas aos 10 anos e 11 meses foi diagnosticado com doença renal crônica secundária à estenose da junção ureteropiélica bilateral.
Ao diagnóstico já apresentava sinais de nefropatia crônica com afilamento do parênquima renal bilateral ao exame de ultrassonografia. Os genitores contaram que aos dois anos a criança já tinha controle esfincteriano diurno. Mas chamava atenção por apresentar poliúria, noctúria e ingerir muita água. Não tinha alteração do jato urinário ou episódios de infecção do trato urinário.
Aos 11 anos e três meses foi submetido a procedimento cirúrgico, pieloplastia com implante de duplo J à direita, com o objetivo de prolongar o tratamento conservador e retardar o início da terapia de substituição renal.
Aos 12 anos e três meses, um ano e quatro meses após o diagnóstico, foi submetido a transplante renal preemptivo com doador vivo relacionado, a mãe.
DiscussãoO caso descrito acima é real e comum, revela o preocupante problema do diagnóstico tardio da DRC em crianças no Brasil. A DRC é definida pela existência de qualquer dano renal, associado a vários graus de redução na taxa de filtração glomerular, conforme representado na tabela 1.1 A DRC em estágio 5, que necessita de terapia de substituição renal (TSR) – diálise ou transplante –, é um problema de saúde pública e há evidências de aumento crescente na incidência e na prevalência dessa condição.2 O número de pessoas que recebem TSR no mundo em 2010 foi estimado em 2,7 milhões de indivíduos, porém também calcula‐se que igual número de pacientes com indicação de TSR não receba tal tratamento por falta de acesso à terapia, principalmente por razões econômicas.3 Calcula‐se ainda que a proporção de pacientes com DRC em estágios 2 a 4 seja muitas vezes superior ao número de pacientes com DRC em estágio 5.4
Classificação dos estágios da doença renal crônica
| Estágio | Descrição | TFG (mL/min/1,73m2) |
|---|---|---|
| 1 | Dano renal com TFG normal/aumentada | >90 |
| 2 | Dano renal com TFG levemente reduzida | 60‐89 |
| 3 | Dano renal com TFG moderadamente reduzida | 30‐59 |
| 4 | Dano renal com TFG gravemente reduzida | 15‐29 |
| 5 | Falência renal | <15 |
Felizmente a proporção de crianças com DRC é muito menor do que a de adultos, porém a DRC em crianças traz consequências devastadoras sobre o crescimento, desenvolvimento ponderoestatural e intelectual, resulta em aumento na morbimortalidade dos indivíduos acometidos.4 A prevalência da DRC pediátrica em nosso meio é inferior à reportada em países desenvolvidos do Ocidente, provavelmente devido à falta de diagnóstico dos casos. Em estudo feito no Brasil foram estimados 1.300 pacientes pediátricos em tratamento dialítico no país em 2012, resulta numa prevalência de 20 casos por milhão de indivíduos com idade compatível (cpmic).5 Na Europa, por exemplo, registros mostram prevalência de 65 cpmic. Nos Estados Unidos, esses dados sobem para 85 cpmic.6
Nos países desenvolvidos a principal causa de DRC na infância são doenças hereditárias, com destaque para as anormalidades congênitas dos rins e trato urinário (CAKUT), que somam de 30% a 60% das crianças com doença renal crônica em TSR.4,6 Em contraste, o principal diagnóstico etiológico que foi atribuído para as crianças em diálise no Brasil foi “causa desconhecida”, com 32% dos casos, que é muito superior ao reportado nos países do ocidente, sugere que o diagnóstico entre nós é formulado de maneira incompleta.5 Outro aspecto que chama atenção é o tempo entre o diagnóstico da DRC e o início da TSR, que no Brasil foi de 14 meses no Sudeste, nove no Centro‐Oeste e nenhum no Nordeste, sugere um retardo no estabelecimento do diagnóstico, o que também deprecia o prognóstico final dessas crianças.5
No caso clínico apresentado acima, o paciente já apresentava ao diagnóstico anemia e baixa estatura, que são sinais de doença detectada tardiamente. O diagnóstico tardio merece discussão, pois, assim como nesse caso, muitos outros podem passar despercebidos sem diagnóstico e gerar prejuízos aos pacientes. O diagnóstico precoce da DRC é uma oportunidade que compete aos pediatras generalistas, e não aos especialistas. Ele possibilita terapias médicas e indicação de procedimentos cirúrgicos quando necessários, que podem retardar a progressão da DRC e a necessidade de início da TSR.
O diagnóstico genético das CAKUT é desafiador e ainda não acessível pelo custo elevado dos exames genéticos e pela complexidade de interações entre os múltiplos genes candidatos a explicar as doenças7 e não será abordado neste manuscrito. Este artigo visa a revisar os sinais e sintomas clínicos associados às CAKUT mais prevalentes na infância, busca fornecer ao pediatra geral elementos para o diagnóstico e tratamento precoces para melhor prognóstico das crianças acometidas.
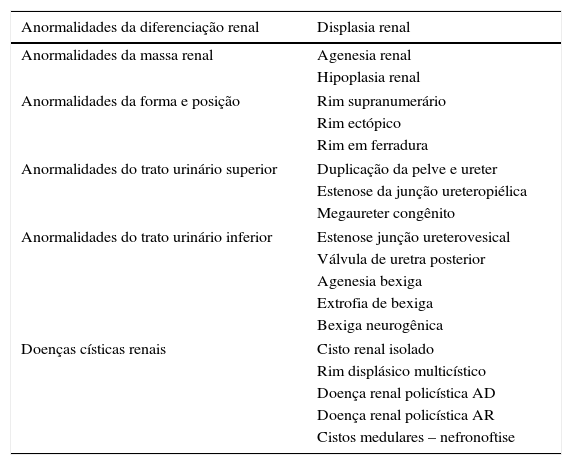
Classificação das anormalidades congênitas dos rins e do trato urinárioAs CAKUT constituem um grupo heterogêneo de patologias do desenvolvimento dos rins e do trato urinário (tabela 2). Em estudo alemão que envolveu 30.940 recém‐nascidos, natimortos e abortamentos, estimou‐se que 7% apresentavam malformações importantes e as CAKUT foram a segunda anomalia mais frequente, com prevalência estimada de 1,6%.8 Em estudo no Brasil com 6.245 necropsias perinatais e pediátricas, os autores observaram que as CAKUT estavam presentes em 2,9% dos casos.9
Anormalidades congênitas dos rins e do trato urinário
| Anormalidades da diferenciação renal | Displasia renal |
|---|---|
| Anormalidades da massa renal | Agenesia renal |
| Hipoplasia renal | |
| Anormalidades da forma e posição | Rim supranumerário |
| Rim ectópico | |
| Rim em ferradura | |
| Anormalidades do trato urinário superior | Duplicação da pelve e ureter |
| Estenose da junção ureteropiélica | |
| Megaureter congênito | |
| Anormalidades do trato urinário inferior | Estenose junção ureterovesical |
| Válvula de uretra posterior | |
| Agenesia bexiga | |
| Extrofia de bexiga | |
| Bexiga neurogênica | |
| Doenças císticas renais | Cisto renal isolado |
| Rim displásico multicístico | |
| Doença renal policística AD | |
| Doença renal policística AR | |
| Cistos medulares – nefronoftise | |
AD, autossômico dominante; AR, autossômico recessivo.
Além de seu papel como principal etiologia da DRC em crianças, entende‐se que as CAKUT representam importante etiologia da DRC também nos adultos. Estudo recente que avaliou 212.930 indivíduos com DRC em idade adulta revelou que pacientes com CAKUT iniciaram TSR aos 31 anos, enquanto indivíduos com outras etiologias necessitaram de TSR apenas aos 61 anos.10 Isso sugere que parte dos adultos com falência renal é portadora de CAKUT, que, como na infância, podem ser reconhecidas e tratadas precocemente, o que expande a importância desse diagnóstico.
Subsídios para o diagnóstico das anormalidades congênitas dos rins e do trato urinárioMalformações associadasAs CAKUT têm causas muito variadas e fatores genéticos, como anormalidades cromossômicas e mutações hereditárias nos genes que regulam o desenvolvimento do aparelho urinário, podem ser causa subjacente da malformação. Além disso, são igualmente importantes as influências ambientais sobre o desenvolvimento fetal. Isso faz com que a relação entre o genótipo e o fenótipo nessas doenças seja complexa. Como ponto de partida para se tentar caracterizar essa complexidade, é importante se considerar que as CAKUT podem aparecer isoladamente ou podem vir associadas a outras malformações ou síndromes.
Em estudo recente feito a partir de 346.841 abortamentos, natimortos ou recém‐nascidos vivos, estimou‐se que 34% dos 1.678 casos de CAKUT observados tinham outras malformações não urinárias associadas. A distribuição dessas associações está representada na tabela 311 e a importância dessas para o pediatra geral é que o encontro dessas anomalias associadas deve ser um ponto de alerta para a busca de casos de CAKUT.
Malformações associadas às Cakut encontradas em 346.381 casos na França de 1979 a 2004 (total de 1.678 Cakut, das quais 563 com outras malformações associadas)
| Anomalias cromossômicas (n=119) | Trissomiado 18 (n=33) |
| Trissomiado 21 (n=26) | |
| Trissomiado 13 (n=24) | |
| Síndrome de Turner (n=11) | |
| Outras (n=25) | |
| Associações não cromossômicas (n=168) | Vacterl (n=56) |
| Meckel‐Gruber (n=36) | |
| Prune‐Belly (n=23) | |
| Outras (n=53) | |
| Associações de base desconhecida (n=276) | |
| Tecido musculoesquelético (n=97) | Polidactilia/sindactilia (n=45) |
| Encurtamento membros (n=30) | |
| Outras (n=22) | |
| Aparelho digestivo (n=91) | Atresia anal (n=38) |
| Vício de rotação (n=29) | |
| Outros (n=24) | |
| Malformações cardíacas (n=88) | CIV (n=42) |
| CIA (n=21) | |
| Outras (n=25) | |
| Sistema nervoso (n=60) | Espinha bífida (n=21) |
| Encefalocele (n=15) | |
| Microcefalia (n=8) | |
| Outras (n=16) | |
| Cabeça e pescoço (n=53) | Dismorfismo facial (n=21) |
| Hipertelorismo (n=19) | |
| Outras (n=13) | |
| Sistema respiratório (n=28) | Hipoplasia pulmonar (n=21) |
| Outras (n=7) | |
| Parede abdominal (n=23) | Onfalocele (n=14) |
| Gastrosquise (n=9) | |
| Outras (n=29) | Lábio leporino (n=23) |
| Hérnia diafragmática (n=6) | |
CIV, comunicação interventricular; CIA, comunicação interatrial.
Modificado de Stoll et al.11
Antecedentes do período antenatal que estão associados às Cakut são mais frequentemente o oligoâmnio, a prematuridade, o baixo peso ao nascer e a restrição do crescimento intrauterino. Estudo feito no Japão mostrou relação entre antecedentes perinatais e CAKUT, sugeriu sinais para se indicar um screening das crianças. Dentre todos dados estudados (14% dos pacientes com CAKUT nasceram prematuros, 18% tinham baixo peso ao nascer, 79% baixo ganho de peso, 18% asfixia, 8% eram oligoâmnios e 12% tinham icterícia), foram encontrados 82% com antecedentes de oligoâmnio e baixo ganho de peso. Isso sugere que esses dois sinais são marcadores para selecionar indivíduos com maior risco para Cakut, com especificidade de 95%.12 Em estudo brasileiro foram avaliados antecedentes preditores de mortalidade associados a Cakut em 29.653 recém‐nascidos entre 1996 a 2006. Nesse estudo revelaram‐se 524 casos de CAKUT, que corresponderam a 17,7 por 1.000 nascidos vivos, e os fatores de risco para mortalidade foram oligoâmnio, CAKUT associada a outras malformações, baixo peso ao nascer e prematuridade.13 Outro antecedente digno de nota é a existência de mais casos de Cakut na família. Em estudo recente revelou que 14% dos 107 casos de CAKUT tinham antecedente familiar com diagnóstico confirmado em ao menos um membro da família.14 Mais um antecedente familiar foi revelado em estudo que envolveu 1994 pacientes com DRC (diagnóstico antes de 21 anos) e 20.032 controles. Baixo peso ao nascer e diabetes materna pré‐gestacional foram significantemente associados a risco de displasia/aplasia renal.15
A literatura é bastante escassa acerca da descrição de sinais e sintomas que alertam para a existência de um comprometimento do sistema urinário. No caso das CAKUT a expressão clínica das malformações pode conceitualmente ser devida a: a) o comprometimento alto do sistema urinário que leva à disfunção renal, que é evolutiva quando ocorre e pode se externar por poliúria, polaciúria, noctúria, infecção urinária, hipertensão arterial sistêmica e baixo ganho de peso14,16–18 ou b) aos sinais de comprometimento do trato urinário inferior com obstrução do trato urinário, como jato urinário fraco, dificuldade para iniciar a micção, bexigoma, enurese não monossintomática, urge/incontinência urinária, disfunção do intestino e da bexiga.19
A controvérsia sobre o uso de episódios de infecção do trato urinário como indicador de CAKUT é bastante acirrada, porém parece que algumas situações configuram maior chance de associação com CAKUT. Idade menor do que dois anos e febre alta (maior do que 38,3°C) são os fatores que podem predizer anormalidades urológicas.20
Finalmente outro dado clínico que é proeminente para permitir o diagnóstico precoce das nefropatias congênitas é a medida da pressão arterial. Sabe‐se que a hipertensão é mais frequente nas crianças nefropatas e inversamente que o controle da pressão arterial tem efeito terapêutico comprovado para reduzir a progressão da DRC em crianças.21 Essas associações reforçam a importância da recomendação de medida da pressão arterial em crianças, que deve ser feita em todas as consultas pediátricas a partir dos três anos e nas crianças menores de três anos quando existir alguma das seguintes situações: a) prematuridade, baixo peso ao nascer ou complicações neonatais que requerem internação em UTI neonatal, b) cardiopatias congênitas, c) infecção urinária, hematúria ou proteinúria, d) doença renal conhecida ou malformações urológicas, e) história familiar de doença renal congênita, f) transplante de órgãos sólidos, g) tumores malignos ou transplante de medula óssea, h) doenças sistêmicas associadas com hipertensão e i) evidência de pressão arterial elevada.22
Alterações de ultrassonografia antenatalO uso da ultrassonografia no acompanhamento da gravidez e também dos neonatos se iniciou no fim dos anos 1970. Atualmente o ultrassom gestacional é parte da rotina médica na maioria dos países, com pelo menos dois exames em cada gestação para acompanhamento do desenvolvimento fetal e identificação de possíveis malformações.23 A ultrassonografia gestacional melhorou substancialmente o diagnóstico e prognóstico das crianças com Cakut, chegou a uma sensibilidade para detecção de uropatias obstrutivas entre 80% e 90%. Em estudo europeu feito em 12 países, a sensibilidade para detecção de malformação renal foi de 81,2%.23
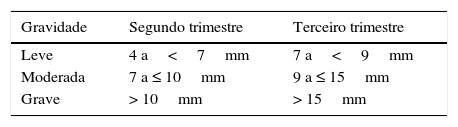
A hidronefrose é o achado no ultrassom mais frequentemente descrito e usado. Hidronefrose antenatal é a presença de um ou ambos os rins com algum grau de dilatação do sistema pielocalicinal no feto. O diagnóstico é feito pela ultrassonografia obstétrica, usa‐se como parâmetro principalmente o diâmetro anteroposterior (DAP) da pelve renal e a presença de caliectasia. Há hidronefrose quando o DAP da pelve está acima de determinados limites, porém ainda não há consenso sobre a definição desses limites. Os valores mais citados, acima dos quais há maior risco de degeneração da função renal ou de necessidade de cirurgia, são os propostos pela Sociedade de Urologia Fetal (SFU),24 que estão resumidos na tabela 4.
O maior valor preditivo positivo (VPP) está no exame feito no terceiro trimestre da gestação. O VPP para valores de DAP >7mm no terceiro trimestre é de 69% versus 49% para DAP>4mm no segundo trimestre.4 A incidência reportada de acometimento fetal varia de 1% a 5% das gestações, depende dos critérios diagnósticos, é mais frequente em meninos (3 a 4/1), predominantemente unilateral e apenas 20% terão significado clínico pós‐natal.
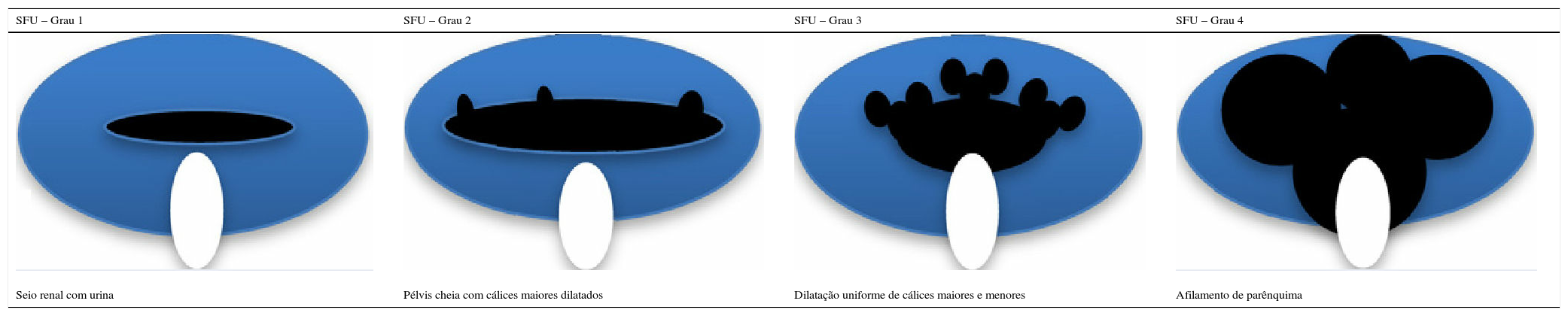
Outro achado ultrassonográfico de interesse para o diagnóstico e prognóstico das crianças com CAKUT é a classificação da hidronefrose proposta pela SFU, que se baseia no grau de dilatação pielo‐calicinal e leva em conta também a integridade do parênquima. O grau 0 representa ultrassonografia normal e os demais graus estão representados na tabela 5.
Representação gráfica e definições da classificação das alterações ultrassonográficas na hidronefrose (adaptado de Timberlake & Herndon25)
| SFU – Grau 1 | SFU – Grau 2 | SFU – Grau 3 | SFU – Grau 4 |
|---|---|---|---|
| Seio renal com urina | Pélvis cheia com cálices maiores dilatados | Dilatação uniforme de cálices maiores e menores | Afilamento de parênquima |




SFU, Sociedade de Urologia Fetal.
Esse sistema de classificação foi desenvolvido para ultrassonografia feita após o nascimento, mas tem sido aplicado na ultrassonografia antenatal e existem evidências de que crianças com hidronefrose de graus 3 e 4 têm risco aumentado de necessidade de cirurgia e deterioração da função renal.25
Qualquer que seja a classificação da hidronefrose, o acompanhamento pós‐natal de crianças com imagens de hidronefrose detectadas no período gestacional mostra resolução em 50% dos casos, caliectasia sem fator obstrutivo incluindo pelve extrarrenal em 15%, estenose da junção ureteropiélica em 11%, refluxo vesicoureteral em 9%, megaureter em 4%, rim multicístico displásico em 2%, ureterocele em 2%, cisto renal em 2% e válvula de uretra posterior em 1%.25–27
Outros achados de imagem obtidos por meio da ultrassonografia gestacional que auxiliam no diagnóstico e prognóstico da criança com CAKUT e também estão associados com deterioração da função renal são: a) o encontro de rim único, situação na qual até 1/3 dos acometidos tem Cakut associada,28,29 b) hidronefrose associada a outras anomalias, quando a probabilidade de DRC aos dois anos é de 15% versus apenas 2% nas crianças com hidronefrose isolada30 e c) hidronefrose bilateral, que por si só confere risco de deterioração na função renal 9,4 vezes maior do que os casos com alteração unilateral.31
Além do aspecto e do grau da hidronefrose, outros achados ultrassonográficos devem ser valorizados, como: a) a dilatação ureteral, presente no megaureter; b) estenose de junção ureterovesical, válvula de uretra posterior e ausente na estenose da junção ureteropiélica; c) o tamanho renal, que pode estar comprometido na hipo/displasia renal ou mesmo na hidronefrose; d) o tamanho vesical e a espessura da parede da bexiga com ou sem formação de trabéculas, alterações que podem estar presentes na bexiga de esforço ou na bexiga hipotônica; e) o volume urinário residual pós‐micção aumentado, associado ao mau funcionamento vesical e f) a ureterocele, geralmente presente na duplicidade ureteral.32
ConclusõesNo presente trabalho buscamos revisar a literatura médica para listar os principais dados clínicos que podem ser úteis na prática pediátrica para identificar precocemente os pacientes portadores de CAKUT. A importância desse diagnóstico deriva do fato de serem as Cakut a causa mais comum de DRC em crianças. No cenário epidemiológico presente, caracterizado por queda da morbimortalidade secundária às doenças infectocontagiosas, as doenças crônicas em geral e a DRC em particular ganham importância na prática clínica e essa tendência deve se acentuar com o tempo.2
Várias evidências recentes apontam que a DRC tem suas origens na vida intrauterina e atualmente sabe‐se que o número de nefros existentes em cada rim pode variar bastante entre indivíduos e que o menor número de unidades funcionais do rim representa risco aumentado de DRC e doença cardiovascular33,34 Tal consideração assume contornos ainda mais importantes nas crianças portadoras de anormalidades do desenvolvimento do sistema urinário, situação na qual a nefrogênese frequentemente é perturbada, resulta em menor número de nefros. Essas constatações antecipam para a idade pediátrica o momento de maior interesse para se fazer o diagnóstico precoce dos pacientes em risco para DRC.
O momento para fazer essa busca diagnóstica deve ser o mais precoce possível, se possível até antes do nascimento. Sabe‐se que mesmo em situações de existência de serviços de seguimento bem organizados a ocorrência de não adesão completa das famílias aos procedimentos necessários para pesquisa diagnóstica é um fenômeno frequente.35
Alguns países buscam o diagnóstico precoce do risco para DRC com programas compulsórios de exames de sedimento urinário ou de imagem em determinados pontos de idade,36,37 mas as análises de custo‐efetividade de tais programas nunca demonstraram definitivamente que essa iniciativa fosse vantajosa.38 Numa situação como essa, a avaliação clínica é um recurso inestimável para triar quais os pacientes devem ser estudados com recursos laboratoriais mais detalhados. Principalmente em países com recursos limitados na área de saúde, tal estratégia parece ser ainda mais acertada. No Brasil, com exceção da Região Sudeste, a causa desconhecida é o mais comum diagnóstico da DRC avançada em crianças,5 o que sugere que há uma lacuna importante a ser preenchida para melhor qualificar o diagnóstico da DRC. É plausível que muitas crianças cheguem ao estágio final da evolução da DRC sem ter tido acesso ao diagnóstico de Cakut, que é a causa mais importante dessa doença no mundo.
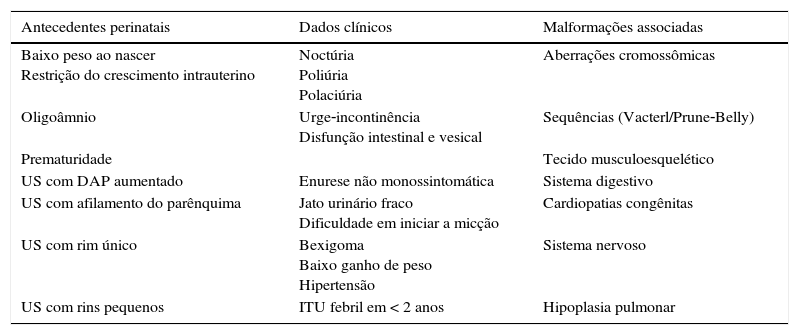
Chama atenção na presente revisão que não existem estudos com metodologia robusta para dar respostas com base em evidências sólidas sobre o tema. A maioria dos estudos revistos é descritiva e unicêntrica e por isso os achados aqui resumidos devem ser considerados como preliminares. Os sinais de alerta para o encontro de crianças portadoras de Cakut estão resumidas na tabela 6.
Sinais de alerta para o diagnóstico de CAKUT em crianças
| Antecedentes perinatais | Dados clínicos | Malformações associadas |
|---|---|---|
| Baixo peso ao nascer Restrição do crescimento intrauterino | Noctúria Poliúria Polaciúria | Aberrações cromossômicas |
| Oligoâmnio | Urge‐incontinência Disfunção intestinal e vesical | Sequências (Vacterl/Prune‐Belly) |
| Prematuridade | Tecido musculoesquelético | |
| US com DAP aumentado | Enurese não monossintomática | Sistema digestivo |
| US com afilamento do parênquima | Jato urinário fraco Dificuldade em iniciar a micção | Cardiopatias congênitas |
| US com rim único | Bexigoma Baixo ganho de peso Hipertensão | Sistema nervoso |
| US com rins pequenos | ITU febril em < 2 anos | Hipoplasia pulmonar |
US, ultrassom, DAP, diâmetro anteroposterior, ITU, infecção do trato urinário.
Acreditamos que, mesmo com as limitações metodológicas dos estudos revistos, as sugestões apresentadas podem ajudar os pediatras a estabelecer hipóteses clínicas para o diagnóstico precoce das anormalidades do desenvolvimento do trato geniturinário valendo‐se exclusivamente de dados clínicos e de informações do ultrassom gestacional, sem metodologias caras e invasivas. Como não existem dados prospectivos para avaliar quais dos sinais e sintomas clínicos aqui relacionados tem maior importância para identificar o risco, acreditamos que, pelo momento, a ocorrência de qualquer um deles deveria ser indicativo para investigar as crianças quanto à possível existência de Cakut o mais precocemente.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
Como citar este artigo: Nogueira PC, Paz IP. Signs and symptoms of developmental abnormalities of the genitourinary tract. J Pediatr (Rio J). 2016;92(3 Suppl 1):S57–63.





