
To discuss the recent literature on paternal obesity, focusing on the possible mechanisms of transmission of the phenotypes from the father to the children.
SourcesA non‐systematic review in the PubMed database found few publications in which paternal obesity was implicated in the adverse transmission of characteristics to offspring. Specific articles on epigenetics were also evaluated. As the subject is recent and still controversial, all articles were considered regardless of year of publication.
Summary of findingsStudies in humans and animals have established that paternal obesity impairs their hormones, metabolism, and sperm function, which can be transmitted to their offspring. In humans, paternal obesity results in insulin resistance/type 2 diabetes and increased levels of cortisol in umbilical cord blood, which increases the risk factors for cardiovascular disease. Notably, there is an association between body fat in parents and the prevalence of obesity in their daughters. In animals, paternal obesity led to offspring alterations on glucose‐insulin homeostasis, hepatic lipogenesis, hypothalamus/feeding behavior, kidney of the offspring; it also impairs the reproductive potential of male offspring with sperm oxidative stress and mitochondrial dysfunction. An explanation for these observations (human and animal) is epigenetics, considered the primary tool for the transmission of phenotypes from the father to offspring, such as DNA methylation, histone modifications, and non‐coding RNA.
ConclusionsPaternal obesity can induce programmed phenotypes in offspring through epigenetics. Therefore, it can be considered a public health problem, affecting the children's future life.
Discutir a literatura recente sobre obesidade paterna, focalizando os possíveis mecanismos de transmissão dos fenótipos do pai para os filhos.
FontesUma revisão não‐sistemática no banco de dados PubMed encontrou poucas publicações com obesidade paterna implicada com a transmissão adversa das características à prole. Artigos específicos sobre epigenética também foram avaliados. Como o assunto é recente e ainda controverso, todos os trabalhos foram considerados independentemente do ano de publicação.
Resumo dos achadosEstudos em seres humanos e animais estabeleceram que a obesidade do pai prejudica seus hormônios, metabolismo e função espermática, que pode ser transmitida à prole. Em humanos, a obesidade paterna resulta em resistência à insulina / diabetes tipo 2 e aumento do nível de cortisol no sangue do cordão umbilical, que aumenta os fatores de risco para doença cardiovascular. Notavelmente, existe associação entre a gordura corporal nos pais e a prevalência de obesidade em suas filhas. Em animais, pais obesos condicionam, na prole, a homeostase glicose‐insulina, lipogênese hepática, hipotálamo / comportamento alimentar, rim, prejudicam o potencial reprodutivo da prole masculina com estresse oxidativo espermático e disfunção mitocondrial. Uma explicação para estas observações (humanos e animais) é a epigenética, considerada a ferramenta básica para a transmissão de fenótipos do pai à prole, como a metilação do DNA, modificações nas histonas, e RNA não codificante.
ConclusõesA obesidade paterna pode induzir fenótipos programados na prole através da epigenética. Portanto, a obesidade paterna pode ser considerada um problema de saúde pública, afetando a vida futura das crianças.
A obesidade tem crescido de forma desordenada, constituindo uma epidemia real descrita como “globesidade”, o que representa um grave problema de saúde pública atualmente.1
Sabemos agora que o risco de desenvolver obesidade e síndrome metabólica (SM) na idade adulta pode ser influenciado pelo período de vida inicial, especialmente através de uma nutrição inadequada disponível para o feto e o recém‐nascido.2,3 “Programação” é como chamamos o processo pelo qual os fatores iniciais da vida podem influenciar a saúde da prole na idade adulta. A programação é considerada um mecanismo essencial para o estabelecimento da obesidade e alterações metabólicas na prole.4,5 Vários modelos são utilizados para compreender os mecanismos associados à programação, em que o ambiente hormonal e metabólico durante o período pré‐natal ou pós‐natal é alterado por mudanças no estado nutricional materno.6–8
A maioria dos estudos epidemiológicos e experimentais centralizou‐se na influência da mãe na saúde da prole. No entanto, experiências recentes com roedores demonstraram o envolvimento do pai afetando a homeostase da glicose e a vida útil das ilhotas pancreáticas na prole feminina.9 Os testes clínicos e em animais desafiaram as ideias convencionais sobre a programação metabólica, sugerindo que algo mais poderia estar envolvido nesse processo através da programação paterna. Estudos recentes agora indicam que a saúde metabólica do pai na concepção também pode afetar a saúde das crianças, com pais obesos mais propensos a gerar uma criança obesa.10
Nesta revisão, relatamos as descobertas recentes e os mecanismos propostos envolvidos na programação do pai na prole.
Estudos humanosEstudos em seres humanos analisaram a relação entre os fatores relacionados ao estilo de vida do pai, os fatores de exposição ambiental e o desfecho de saúde da prole no início da vida e posteriormente, sugerindo que os efeitos paternos podem desempenhar um papel significativo na patogênese das doenças crônicas da prole na vida adulta (por exemplo, resistência à insulina e diabetes tipo 2). Mais de 60% de todos os adultos são classificados como tendo sobrepeso ou obesidade na maioria das sociedades ocidentalizadas e, à medida que a prevalência da obesidade aumenta, é responsável por uma proporção cada vez maior do fardo geral da doença.9,11
Há evidências claras de que os fatores nutricionais do pai desempenham um papel significativo na saúde da prole. Por exemplo, há uma correlação entre quantidades absolutas e relativas de gordura corporal paterna e os mesmos parâmetros em suas filhas com idade entre 4,8 e 8,9 anos.12 Além disso, o índice de massa corporal do pai (IMC) pode modular o fenótipo da prole de maneira sexo‐dependente. Nos quadros de um estudo de coorte familiar (899 trios consistindo de pais e prole), o IMC paterno correlacionou‐se com o peso ao nascer, o diâmetro biparietal, a circunferência da cabeça, o diâmetro abdominal, a circunferência abdominal e o diâmetro torácico apenas nos recém‐nascidos do sexo masculino.12 Os pais13 ou avôs14 expostos à sobre alimentação ou restrição alimentar no período de idade de 9 a 12 anos predeterminam longevidade reduzida na prole masculina. A segunda geração de descendentes desses avôs teve um risco quatro vezes aumentado de mortalidade por diabetes.15
No norte da Suécia, o seguimento de três gerações demonstrou que o consumo excessivo de alimentos de um avô está associado à redução da capacidade de sobrevivência14 e ao aumento do risco de diabetes15 em seus netos. O início precoce do tabagismo do avô também está relacionado ao aumento do IMC do neto.16 Essa evidência indica que os fatores nutricionais do pai, não apenas antes da concepção, mas também até mesmo na puberdade do pai, podem afetar a prole de maneira sexo‐dependente.17 Além disso, existe uma interação entre genes parentais e fatores ambientais parentais que têm um efeito sobre o fenótipo da prole.18 A interação gene‐meio ambiente torna‐se ainda mais complicada, pois também se sabe que o status socioeconômico de um indivíduo parece ter efeitos opostos sobre a obesidade em países pobres e ricos.19
No pai, o desequilíbrio calórico imposto pelas escolhas de estilo de vida, incluindo o alto consumo de alimentos e a baixa atividade física, são fatores a serem considerados em estudos de programação. As modificações epigenéticas podem ocorrer durante a vida de muitos indivíduos dentro de uma população e, assim, serem transmitidas imediatamente para um grande número de descendentes na próxima geração, ao contrário dos eventos genômicos que se espalham lentamente por uma população.20 É provável que mudanças nas circunstâncias em dentro do indivíduo ou ao longo de várias gerações possam recrutar alelos silenciosos de volta ao genoma ativo e contribuir para a reversibilidade de mudanças adaptativas ou adquiridas. Um estudo recente em homens obesos mostrou alterações no microRNA circulante (miRNA) que têm como alvo o VEGF (fator de crescimento endotelial vascular), proteínas quinases de mitogênio ativado no tecido adiposo, que foi reversível após a perda de peso.21
O IMC do pai durante a concepção foi associado ao desenvolvimento fetal da prole masculina, mas não da prole feminina. O IMC do pai correlacionou‐se significativamente com o peso ao nascer e o diâmetro biparietal perinatal, a circunferência da cabeça, o diâmetro abdominal, a circunferência abdominal e o diâmetro torácico medido na prole masculina. Não houve correlações significativas entre o IMC paterno e esses parâmetros na prole feminina. O nível de cortisol do sangue do cordão umbilical também foi associado ao IMC do pai apenas na prole masculina. Os autores concluíram que um efeito transgeracional sexo‐específico do IMC do pai na secreção fetal de cortisol pode representar um fator de risco para doenças cardiovasculares em prole masculina na vida adulta.12 Além disso, o aumento do IMC do pai está associado à diminuição do desenvolvimento do blastocistos e taxas de nascidos vivos após a fertilização in vitro.22 No pai obeso (IMC> 25kg/m2), detectou‐se uma elevação nas espécies reativas de oxigênio no esperma, aumento dos níveis de neopterina no líquido seminal (um marcador de ativação de macrófagos do trato reprodutor), diminuição da contagem de espermatozoides e níveis de testosterona sérica e aumento do estradiol sérico.23
Estudos em animaisModelos animais de obesidade masculina estão sendo utilizados para avaliar o impacto da programação paterna na prole e analisar a função espermática do pai obeso. Os modelos animais são importantes devido às dificuldades em separar os efeitos da composição genética paterna das exposições ambientais do pai sobre a prole,10 bem como agrupar e interpretar dados de estudos humanos. A melhor compreensão dos mecanismos de programação paterna pode ajudar as intervenções para minimizar os efeitos adversos sobre a prole.24
Uma programação paterna (quando os pais obesos levam ao distúrbio da homeostase glicose‐insulina na prole feminina) foi inicialmente descrita em animais.9 Pais obesos também programam a lipogênese e a beta‐oxidação no fígado25 e o hipotálamo da prole (a inflamação do hipotálamo foi encontrada na prole, com o aumento das expressões de IL (interleucina)‐6 e TNF (fator de necrose tumoral).6
Os pais obesos também alteraram o rim na prole, com danos tubulares e perda da borda em escova tubular, mas não danos glomerulares. O gene Acat1 (gene da colesterol‐aciltransferase‐1), envolvido na entrada de ácido graxo para a beta‐oxidação nos túbulos, mostrou up‐regulação na prole.26
O desenvolvimento de células germinativas masculinas de mamíferos é suscetível a dano em diferentes momentos em doenças específicas da prole na vida adulta: desenvolvimento embrionário, primeira infância e idade pré‐púbere, e preconcepção e espermatogênese na idade adulta.27‐29 A obesidade do pai afeta negativamente o potencial reprodutivo da prole masculina, não só alterando a função, qualidade e composição molecular do esperma, mas também aumentando o estresse oxidativo do esperma, contribuindo para o dano ao DNA e a disfunção mitocondrial.30
Muitos estudos em modelos animais concentram‐se nos fatores adversos que influenciam a exposição do pai do período pré‐púbere à preconcepção. Portanto, é mais plausível considerar que o epigenoma sofre reprogramação em embriões pré‐implantação e células germinativas primordiais.31
Tem sido sugerido que as exposições ambientais em modelos animais durante esse período induzem efeitos intergeracionais e transgeracionais através do epigenoma do esperma.32 Os dados demonstram que muitos efeitos metabólicos observados na primeira geração continuam na segunda geração com fêmeas F1 produzindo machos F2 com aumento de adiposidade. A obesidade do pai modificou a expressão de vários microRNAs, concomitante com alterações no conteúdo de microRNA do esperma e uma redução na metilação global do DNA das células germinativas.32
Desde o momento em que o blastocisto é formado, a pré‐implantação acabou, e uma nova etapa começa – o desenvolvimento dentro do útero. Depois de entrar nas gônadas, as células germinativas primordiais se convertem em gonócitos e em modelos de camundongos ocorre a diminuição dos níveis de metilação do DNA.33 As responsabilidades primárias nesse tempo de janela abrangem a expressão do gene célula‐específico, a diferenciação de tecidos e os epigenomas tecido‐específicos.27
Pais camundongos obesos tiveram retenção de H3 e impressões genômicas no esperma e diferenças na expressão de mRNA hepático de vários genes relacionados à síntese de gordura na prole. Observaram‐se diferenças na expressão hepática de Mt1 e Mt2 (metalotioneína‐1 e ‐2), Fasn (ácido graxo sintase), P450 citocromo oxidoredutase34 e Acaca (acetil‐CoA carboxilase‐α) com a idade de 24 semanas. O pai obeso também aumentou a ocupação da histona H3 nos promotores de genes responsáveis pelo desenvolvimento embrionário e aumento do H3K4me1 (monometilação da lisina 4 na histona H3) em genes responsáveis pela regulação da embriogênese nos espermatozoides. No total, os achados sugerem que a exposição dietética pode modular a composição de histonas em genes envolvidos no processo de desenvolvimento.35
EpigenéticaOs mecanismos que explicam como o pai pode afetar o desenvolvimento da prole ainda estão em debate. A epigenética é a principal ferramenta para a transmissão de fenótipos paternos à prole, porque os estímulos nutricionais transitórios em estágios críticos da ontogênese podem ter influência na expressão de vários genes através de mudanças na conformação da cromatina e na acessibilidade dos fatores de transcrição.36
Portanto, o termo epigenético (prefixo grego epi‐ (¿π¿): sobre, fora de, ao redor), proposto por Conrad Waddington, é descrito como um “processo de desenvolvimento do fenótipo a partir do genótipo”.37 Em outras palavras, a epigenética é qualquer modificação transmissível e reversível na expressão de um gene sem alteração estrutural na sequência do DNA.38 Diferente da variação genética da linha germinal, que permanece inalterada em todas as células do corpo, a modificação epigenética é dinâmica e varia entre os tecidos em resposta a uma série de estímulos ambientais, incluindo aqueles que direcionam a diferenciação tecidual durante o desenvolvimento e o crescimento e os graves riscos que provocam uma resposta adaptativa das células.39
Alguns marcos epigenéticos durante a espermatogênese podem continuar durante o desenvolvimento embrionário. As exposições ambientais (dieta, estilo de vida e outras exposições) que ocorrem na gametogênese masculina podem causar alterações epigenéticas irreversíveis e consequências fenotípicas expressas na prole.40 Os processos epigenéticos também modulam os efeitos através da regulação da transcrição, devido a vários processos, tais como a metilação do DNA, alterações das histonas e a transcrição do RNA não‐codificante (miRNA, por exemplo).41,42
As modificações epigenéticas que atuam sobre a capacidade de plasticidade celular, preparam o indivíduo para o ambiente extrauterino e podem potencializar uma vantagem de sobrevivência, regulando os genes diferenciais que codificam proteínas envolvidas no metabolismo energético e na adipogênese.43 No entanto, diante de uma condição metabólica deletéria como a obesidade e alterações metabólicas relacionadas, essas modificações podem ser exacerbadas ou silenciadas, especialmente a programação das células germinativas, que constituem a perpetuação e a transmissão fenotípica da espécie.44
Hipóteses recentes consideram que alguns padrões alimentares paternos residem em espermatozoides carreando informações epigenéticas.27 O “epigenoma” do esperma era tradicionalmente considerado insignificante, pois postulava‐se que seu perfil de metilação do DNA era apagado imediatamente após a fertilização. No entanto, nos últimos anos, houve um aumento no número de casos relatados de aparente herança epigenética através da linha germinal masculina, sugerindo que este epigenoma pode transmitir informações entre gerações.28,40
O desenvolvimento de embriões de rã derivados de espermatozoides e espermátides permite observar que o esperma, além da transferência do DNA, também contribui para a informação epigenética necessária para a expressão adequada do gene embrionário, representando a chave para a informação epigenética.45
MetilaçãoA metilação do DNA é uma das reações químicas que ocorrem mais frequentemente em eucariotas, como plantas, fungos, invertebrados e vertebrados.34 Esta modificação química é caracterizada pela adição de um grupo metil na posição C5 do anel de citosina, catalisada pelas metiltransferases de DNA, levando à formação de 5‐metilcitosina.46,47 A frequência de 5‐metilcitidina é inferior a 1% do número total de nucleotídeos no genoma.48 Esse processo de metilação atua no desenvolvimento embrionário normal, inativação do cromossomo X, regulação genética, impressão genômica e modificações da cromatina.49
A maior parte das metilações do DNA ocorre em regiões chamadas ilhas CpG, que correspondem às regiões genômicas com mais de 1.000 pares de bases de comprimento e com muitos dinucleotídeos CG, com cerca de 55% dessas ilhas localizadas nas regiões promotoras de aproximadamente 40% dos genes de mamíferos. Estas ilhas CpG (citosina‐fosfato‐guanina) são mantidas não‐metiladas, exceto em genes de impressão ou quando localizadas no cromossomo X inativo.50,51 Portanto, as ilhas CpG localizadas na região promotora de genes constitutivos (housekeeping) e genes reguladores do desenvolvimento com distribuição densa de citosina e guanina são resistentes à metilação do DNA.52 As ilhas CpG, que permitem a ligação de proteínas e enzimas, iniciam a cascata de transcrição. Em contraste, as ilhas CpG metiladas estão relacionadas ao silenciamento transcricional.53
A hipermetilação das regiões promotoras ricas em dinucleotídeos tem um papel significativo na perda de expressão gênica.54 Normalmente, a hipometilação do DNA desencadeia um aumento na expressão gênica, enquanto a hipermetilação diminui a expressão dos genes‐alvo.55 Além disso, os fatores de transcrição não reconhecem e se ligam aos locais de início da transcrição devido à modificação da citosina em 5‐metilcitidina. Tal é o caso de AP‐2 (proteína adipócita 2), cMYC / MYN (cMYC / homólogo murino de max), CREB (proteína de ligação ao elemento de resposta de monofosfato de adenosina cíclica), E2F (fator E2) e NF‐KB (fator nuclear‐ κB). No entanto, esses locais de ligação podem ser ocupados por outras proteínas tais como MeCP‐2 (proteína de ligação de metil‐CpG 2), MBD (proteína de domínio de ligação de metil‐CpG) 1, MBD2, MBD3 e MBD4, que se ligam a citosinas metiladas e estimulam a condensação da cromatina, inativando o gene.46,56
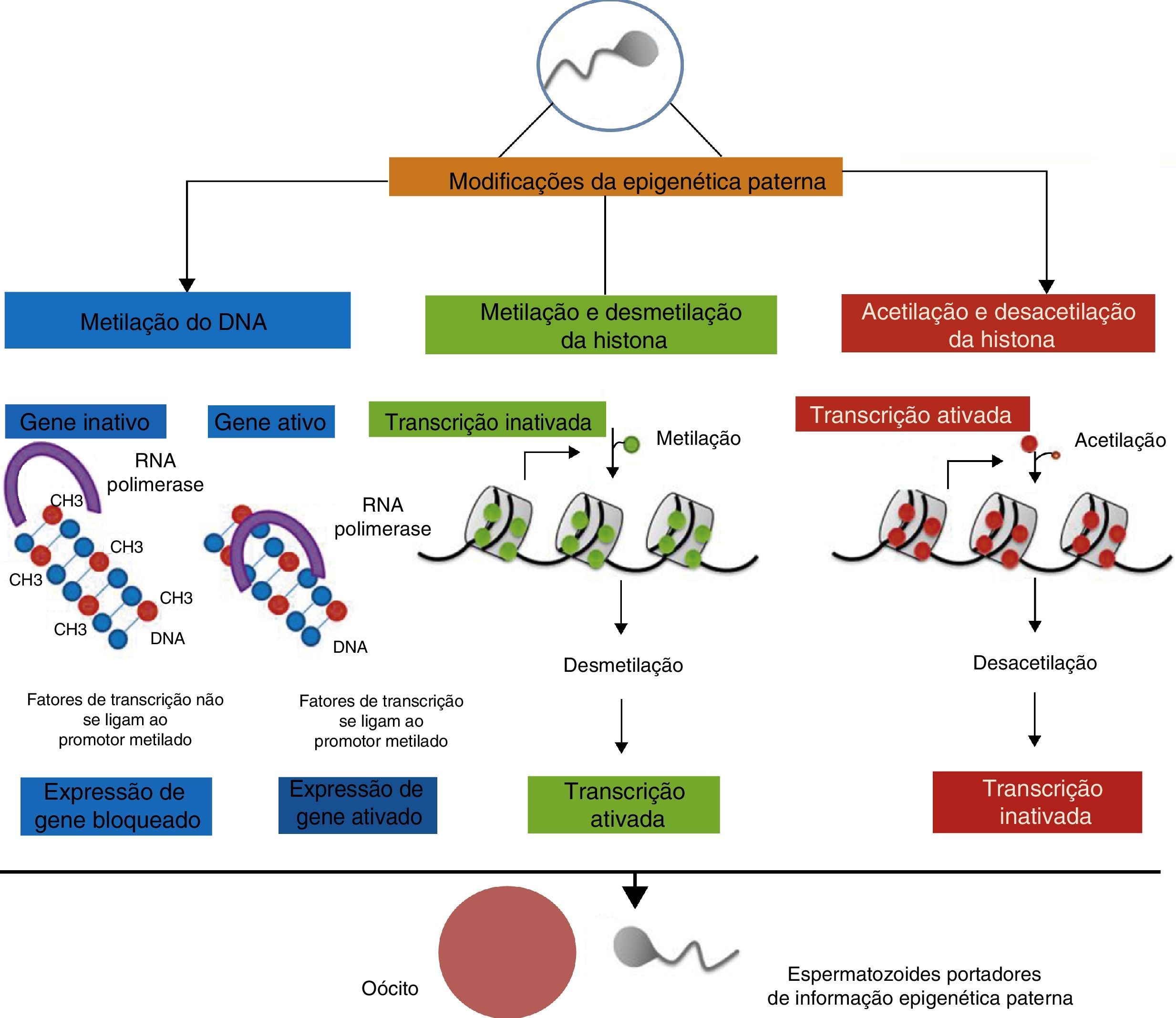
As enzimas responsáveis pela adição de um grupo metila às moléculas de citosina pertencem à família de metiltransferases de DNA (DNMTs), incluindo DNMT (DNA metiltransferase) ‐1, DNMT3A, DNMT3B e suas isoformas, e DNMT3L.47,57 DNMT1 é a principal responsável pela manutenção dos padrões de metilação do DNA durante a mitose. Além disso, DNMT (regiões diferencialmente metiladas) ‐s3 é responsável pela metilação de novo de moléculas de DNA recém‐sintetizadas e é mais importante durante os primeiros estágios do desenvolvimento embrionário e pode ser o processo envolvido na programação paterna para a transmissão fenotípica.58,59 Uma visão esquemática das principais modificações epigenéticas paternas pode ser vista na figura 1.
 favorece a condensação da cromatina, dificultando o acesso às proteínas reguladoras que promovem a transcrição. No entanto, as histonas não‐metiladas garantem a cromatina descondensada, apoiando a transcrição do gene. Em contraste, a acetilação de histonas abre a cromatina, permitindo o acoplamento da maquinaria transcricional.")
Epigenética da programação do pai sobre a prole. As alterações metabólicas do pai relacionadas à obesidade, podem resultar em modificações na informação genética contida no espermatozoide. A metilação do DNA em regiões promotoras específicas frequentemente evita a transcrição genética ao inativar o gene em questão. Quanto às modificações das histonas, a hipermetilação (dependendo da histona e do aminoácido) favorece a condensação da cromatina, dificultando o acesso às proteínas reguladoras que promovem a transcrição. No entanto, as histonas não‐metiladas garantem a cromatina descondensada, apoiando a transcrição do gene. Em contraste, a acetilação de histonas abre a cromatina, permitindo o acoplamento da maquinaria transcricional.
Em um processo contrário, a desmetilação do DNA é também um importante componente epigenético da transcrição gênica e programação epigenética, que ocorre através de várias reações enzimáticas que fazem mediação da oxidação da 5‐metilcitosina em 5‐hidroximetilcitosina.60 A presença de 5‐hidroximetilcitosina em regiões promotoras resulta em aumento da transcrição, indicando um papel na regulação em longo e curto prazo da expressão gênica.61
Modificações da histonaAs modificações da histona são outras ações biológicas que regulam a expressão gênica. As histonas são proteínas básicas encontradas nos núcleos celulares eucariotas, que ajudam na compactação do DNA nos nucleossomas, os blocos de ligação da cromatina.62 A cromatina deve ser remodelada, modificando a acessibilidade das ferramentas de transcrição do DNA para controlar o processo de transcrição.63 Eventos como acetilação, metilação, fosforilação e ubiquitinação, geralmente ocorrem na cauda das histonas que se estendem a partir do centro dos nucleossomos.64 Outras modificações químicas também alteram as histonas, tais como a glcNAcilação de histona lisina (acilação da glucosamina), butirização, malonilação e crotonilação.65,66
As histonas 3 e 4 (H3 e H4, respectivamente) são comumente estudadas e a acetilação é a principal modificação epigenética considerada que ocorre na lisina e arginina e neutraliza a carga positiva de resíduos básicos. As enzimas histona acetilase adicionam grupos acetil aos resíduos de histona lisina, e acredita‐se que as histonas acetiladas tenham uma afinidade reduzida entre DNA e histonas, deixando a cromatina em estado relaxado (eucromatina) e transcricionalmente ativa.66
Em contraste, a histona desacetilase remove os grupos acetil, mais condensados e evita a expressão gênica.67 Essas modificações químicas alteram a interação entre DNA e histonas, alterando o grau de dobragem da cromatina e a atividade gênica.68 Portanto, a heterocromatina está relacionada com hipoacetilação para H3 e H4, e di ou trimetilação do nono resíduo de lisina em H3 (H3K9me2 ou H3K9me3).69
RNA não codificante (microRNA)Além da metilação do DNA e da modificação das histonas, o RNA do esperma pode ser um regulador epigenético. Os espermatozoides contêm um array de ambos os mRNA70 e RNA não codificante, incluindo o miRNA.71 A maior parte deste RNA é entregue ao oócito. No entanto, o papel do miRNA na fase inicial da pré‐implantação embrionária ainda está sendo debatido.
A evidência do efeito biológico direto dos miRNAs no período pré‐implantação é apoiada por observações de camundongos com lesão cromossômica no gene Dicer. A perda de processamento enzimático do miRNA Dicer em oócitos leva à letalidade no início do desenvolvimento, onde os zigotos não conseguem sobreviver à divisão em primeiro lugar.72 Portanto, isso sugere a interferência do miRNA no desenvolvimento do zigoto.
O miRNA é uma parte importante do grupo não codificante tão pequeno. Esses componentes possuem moléculas de RNA de aproximadamente 21nt de comprimento que reprimem seu mRNA alvo.73 No sexo masculino, eles estão no núcleo dos espermatozoides, mantendo o DNA conectado às histonas durante a espermiogênese e no desenvolvimento embrionário precoce.74 O miRNA regula várias funções biológicas, demonstrando influência na inativação epigenética de genes e na proteção do DNA contra vírus e transposons.75
De uma maneira geral, o miRNA em animais está localizado principalmente dentro dos introns de genes de RNA codificantes ou não codificantes de proteínas,76 sendo produzidos por transcrição da RNA polimerase II. Nos seres humanos, o miRNA parece ser sintetizado pela RNA polimerase III.77 Nos últimos anos, a super‐ ou a sub‐expressão do miRNA tem sido associada ao desenvolvimento de doenças, mas os mecanismos e ações ainda são ambíguos.78
Epigenética na programação paternaVários estudos em animais e humanos demonstraram a influência da dieta do pai sobre o fenótipo da prole através do epigenoma.
Camundongos machos alimentados com uma dieta rica em gordura geraram uma prole feminina com homeostase glicose‐insulina deficiente, associada à expressão alterada em um dos 642 genes de ilhotas pancreáticas e ao gene hipometilado IL13ra2 (receptor de interleucina 13 alfa 2).9 As alterações foram encontradas recentemente nos transcriptomas de tecidos adiposos retroperitoneais e de ilhotas pancreáticas na prole feminina. No tecido adiposo retroperitoneal, 5.108 genes foram diferencialmente expressos devido à dieta rica em gordura de um pai, cujas funções estão relacionadas à resposta mitocondrial e celular ao estresse, sinalização da telomerase, morte e sobrevivência celular, ciclo celular, crescimento celular e proliferação e câncer.79
Em um modelo de camundongo de dieta do pai rica em proteínas, a prole mostrou uma metilação elevada em PPAR (receptor ativado por proliferador de peroxissoma)‐alpha no fígado, um gene envolvido na formação das primeiras enzimas para a oxidação de lipídeos nas mitocôndrias, sendo um regulador lipídico essencial.80 Além disso, a resistência à insulina do pai alterou o estado de metilação de vários genes sinalizadores de insulina na prole aumentando a susceptibilidade ao diabetes na prole através de alterações epigenéticas no gameta. Tais alterações incluem os genes Pik3r1 (subunidade 1 reguladora de fosfoinositídeo 3‐quinase), Pik3ca (polipeptídio alfa catalítico de fosfinosideto‐3‐quinase), Ptpn1 (proteína tirosina fosfatase, não receptora do tipo 1) e Pik3ca no esperma.81 A dieta rica em gordura do pai em camundongos levou a um aumento no RNA transportador (tRNA) no esperma como uma chave epigenética hereditária influenciada pela dieta do pai e relacionada à insuficiência metabólica na prole, causando intolerância à glicose e resistência à insulina.82
A obesidade paterna inicia distúrbios metabólicos em duas gerações de camundongos, alterando o perfil transcricional do testículo e o conteúdo de miRNA do esperma. O conteúdo diferencial de miRNAs canônicos no esperma sugeriu desregulação na espermatogênese, desenvolvimento embrionário e função metabólica. Além disso, a dieta rica em gordura do pai afetou o status metabólico da prole através de alterações epigenéticas nos genes da adiponectina e leptina por duas gerações.83 Digno de nota, um aumento no miR‐29 (microRNA 29) foi associado à uma diminuição na metilação de elementos repetidos na linha germinal masculina.32 Além disso, 13 miRNAs espermáticos foram modulados pela dieta rica em gordura de um pai e transferiram essa carga de miRNA alterada para o embrião na fertilização, alterando sua trajetória de crescimento e afetando o fenótipo da prole adulta.84
Nos recém‐nascidos, havia uma associação entre a obesidade pré‐concepção e os perfis de metilação do DNA na prole, particularmente nas DMRs (regiões diferencialmente metiladas) do gene do fator de crescimento semelhante à insulina‐2 (IGF‐2) imprintado. A hipometilação na DMR do IGF2 na prole foi associada à obesidade do pai.85
É concebível que os reguladores epigenéticos adicionais ou novos (como os príons) estejam presentes no esperma, ou que a qualidade do esperma seja afetada pela dieta, ou que o meio‐ambiente possa causar as alterações genéticas (embora seja importante enfatizar que cepas de ratos consanguíneos foram usadas nesse estudo).80 Além disto, o dano ao DNA do esperma induzido pela radiação γ mostrou ser hereditário, com descendentes exibindo danos ao DNA do esperma de maneira similar.86
A modificação epigenética é um processo contínuo, e algumas mudanças podem ser reversíveis.20 Estes dados em conjunto revelam o impacto da programação paterna (particularmente através de manipulações nutricionais) sobre a vida futura da prole, influenciada principalmente pela transmissão do fenótipo através do processo epigenético.
Considerações finaisDados de estudos epidemiológicos e em animais fornecem evidências de que a alimentação paterna e as condições de saúde do pai podem programar as gerações posteriores. Assim, a mãe não é a única responsável pela saúde da prole. O pai compartilha a responsabilidade de fornecer uma impressão epigenética esperma‐específica para o oócito, afetando a trajetória do desenvolvimento embrionário e a saúde da prole adulta. Embora o papel da influência paterna possa ser claramente identificado, nosso conhecimento de reprogramação, alteração e possível prevenção de efeitos paternos permanece limitado.
Até à presente data, os estudos humanos não estão progredindo da mesma forma que nos animais; há pouca informação sobre o mecanismo e a contribuição da programação paterna na saúde da criança, uma vez que os homens são amplamente utilizados como controle para pesquisas em mulheres. Sendo assim, considerando que a obesidade do pai também pode ser um problema de saúde pública, é importante melhorar os estudos epidemiológicos para avaliar o papel exato da saúde do pai no esperma e na saúde das crianças a fim de propor novas intervenções para a restauração.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
AgradecimentosNosso laboratório (www.lmmc.uerj.br) é patrocinado pelo Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, subsídio n° 302.154 / 2011‐6 para CAML e n° 306.077 / 2013‐2 para MBA) e Fundação Carlos Chagas Filho de Amparo à Pesquisa do Rio de Janeiro (FAPERJ, subsídio n° 102.944 / 2011 para CAML, # 103.062 / 2011 para MBA).
Como citar este artigo: Ornellas F, Carapeto PV, Mandarim‐de‐Lacerda CA, Aguila MB. Obese fathers lead to an altered metabolism and obesity in their children in adulthood: review of experimental and human studies. J Pediatr (Rio J). 2017;93:551–9.





