
To evaluate, in a sample of patients with disorders of sex development (DSD), data related to the age at referral and their correlation with the initial complaints, gender at referral, defined gender after diagnosis and etiological diagnosis.
MethodsRetrospective review of the age at the first consultation and the reason for it, initial social gender and gender after the diagnosis, karyotype and etiological diagnosis of all cases treated at a DSD outpatient clinic between 1989 and 2016. Cases that did not involve DSD and DSD diagnoses that do not usually involve ambiguous genitalia, thus not requiring specialized monitoring, were excluded.
ResultsOf the 1793 treated cases, 1139 were diagnosed with some type of DSD. This study excluded 430 cases (272 with Turner's syndrome, 66 with Klinefelter syndrome, and 92 with pure gonadal dysgenesis), thus a total 709 individuals were included. Of these, 82.9% were referred due to ambiguous genitalia; only one‐quarter were still in the first month of life, and 6.6% were referred due to pubertal delay, with most of them aged 10 years or older. Of these patients, 68.6% had a diagnosis of XY DSD, 22.4% of XX DSD, and 9% of sex chromosome abnormalities.
ConclusionsThis study presents the largest series in the literature of patients with DSD treated in a single center. The time of referral of the majority of patients with ambiguous genitalia fell short of the ideal, and milder cases of ambiguous genitalia and many with pubertal manifestations were referred even later. The results reinforce the importance of continuing education for professionals who will have the first contact with these patients, mainly pediatricians and neonatologists.
Avaliar em uma amostra de pacientes com distúrbios da diferenciação do sexo (DDS), dados relacionados à idade, ao encaminhamento e sua correlação com as queixas iniciais, ao sexo ao encaminhamento e ao sexo final e diagnóstico etiológico.
MétodosRevisão retrospectiva da idade por ocasião da primeira consulta e motivo dela, sexo social inicial e após definição do diagnóstico, cariótipo e diagnóstico etiológico de todos os casos atendidos em um ambulatório especializado em DDS entre 1989 e 2016. Foram excluídos casos que não compreendiam DDS e diagnósticos de DDS que não cursam comumente com ambiguidade genital, não necessitam de acompanhamento especializado.
ResultadosDos 1.793 casos atendidos, 1.139 foram diagnosticados com algum DDS. Excluíram‐se 430 (272 síndrome de Turner, 66 síndrome de Klinefelter e 92 disgenesia gonadal pura), totalizando 709. Desses, 82,9% foram encaminhados por ambiguidade genital, somente um quarto ainda no primeiro mês de vida e 6,6% por atraso puberal, a maioria com 10 anos ou mais; 68,6% tiveram diagnóstico de DDS XY; 22,4% DDS XX e 9% de anomalias dos cromossomos sexuais.
ConclusõesEste estudo apresenta a maior casuística na literatura de pacientes com DDS atendidos em um único serviço. O momento de encaminhamento da maioria dos pacientes com ambiguidade genital foi aquém do ideal e casos mais leves de ambiguidade e muitos com manifestações puberais foram encaminhados ainda mais tardiamente. Os resultados reforçam a importância do ensino continuado a profissionais que terão o primeiro contato com esses pacientes, principalmente pediatras e neonatologistas.
Desde o Consenso de Chicago, publicado em 2006, os distúrbios da diferenciação do sexo (DDS) são definidos como condições congênitas em que o desenvolvimento do sexo cromossômico, gonadal ou anatômico é atípico.1 Dessa forma, são englobadas condições de etiologia e manifestações clínicas diversas e raras em sua maioria.
Embora se trate de condições resultantes de defeitos genéticos predominantemente congênitos, nem sempre os DDS terão manifestação ao nascimento, cabe primeiramente ao neonatologista, ao pediatra geral ou mesmo ao clínico considerar esse diagnóstico.1–3
Nos casos de alterações já observadas ao nascimento, a criança pode apresentar diferentes graus de ambiguidade genital.1–4 Já durante a infância ou a adolescência, deve‐se suspeitar de DDS em casos de puberdade de início atrasado para a idade ou aquelas com progressão diferente da esperada.1,2,4–8 Por fim, no adulto, a falência gonadal precoce e a infertilidade são quadros que devem levar à suspeição de um DDS.1,2
Apesar de sua raridade, os DDS não caracterizam somente uma doença orgânica, mas também um agravo de caráter social, pela possível dificuldade de definir o sexo biológico e pelos equívocos que podem ser gerados pelo desconhecimento da diferença dos conceitos de sexo biológico, definido a partir dos órgãos sexuais da criança, identidade de gênero, o gênero psicológico com o qual a pessoa se identifica e identidade sexual, o sexo ao qual a pessoa tem voltado seu desejo e afetividade.9
Portanto, preconiza‐se que os DDS sejam atendidos em centros especializados, com equipe multidisciplinar experiente e preparada para lidar não somente com a investigação diagnóstica e a conduta terapêutica, mas também com o apoio psicológico exigido durante o processo.1,2
São escassos os dados na literatura sobre a frequência das diversas etiologias de DDS nos diferentes grupos etários (recém‐nascido e lactente, pré‐escolar e escolar, adolescente e adulto), bem como a respeito de queixas clínicas iniciais, resultados dos cariótipos, sexo de criação e sexo final desses pacientes. Portanto, os objetivos deste estudo foram avaliar, de acordo com a faixa etária, a etiologia dos casos de DDS atendidos em um centro especializado, assim como as queixas iniciais, os cariótipos, os sexos inicial e final e suas relações, levou‐se em conta o momento do encaminhamento desses pacientes.
MétodosA casuística foi composta por indivíduos com DDS encaminhados a um centro especializado entre janeiro de 1989 e dezembro de 2016, em diferentes faixas etárias, e que já tinham o diagnóstico etiológico confirmado no momento do presente estudo.
Os dados obtidos a partir do levantamento dos prontuários dos pacientes foram: idade e sexo social por ocasião da primeira consulta, queixa ao ser encaminhado ao serviço, cariótipo, diagnóstico etiológico confirmado e sexo final, definido ao fim da investigação etiológica. A queixa principal foi separada em ambiguidade genital, atraso puberal, hipogonadismo, ginecomastia, amenorreia, infertilidade, dismorfismos ou malformações e aconselhamento genético. O diagnóstico etiológico foi confirmado segundo a classificação sugerida pelo Consenso de Chicago.1
Foram excluídos os casos que não confirmaram diagnóstico de DDS e os casos de síndrome de Turner, síndrome de Kinefelter e disgenesia gonadal pura por, em geral, não se apresentarem com ambiguidade genital e serem quadros que podem ser diagnosticados e avaliados em serviços de endocrinologia não especializados em DDS.
O estudo foi aprovado pelo Comitê de Ética em Pesquisa (CAAE 97392018.0.0000.5404).
A análise estatística foi feita com o programa SPSS (Statistical Package for the Social Sciences, Inc., Chicago, IL, USA) versão 20.0. Os dados foram apresentados em tabelas com frequência absoluta e relativa de cada motivo da consulta e diagnóstico etiológico, divididos pelas diferentes faixas etárias: 0?1 mês, 2?12 meses, 13?119 meses, 120?239 meses e maior do que 240 meses.
ResultadosTodos os pacientes atendidos no serviço entre janeiro de 1989 e dezembro de 2016 tiveram seus dados localizados e analisados, total de 1.793 indivíduos. Desses, 1.139 casos tiveram diagnóstico final confirmado de uma etiologia de DDS. Os 654 indivíduos cujo diagnóstico final não foi de DDS compreenderam casos de malformação genital congênita isolada (42), malformações congênitas múltiplas, inclusive a do genital (17), pacientes com sinais sugestivos de síndrome de Turner ou de Klinefelter, porém sem alterações de cariótipo (392 e 142, respectivamente), atraso puberal não relacionado a DDS (29) e transgeneridade (32). Esses casos foram excluídos da análise e considerados como erro de encaminhamento ao ambulatório (36,4%).
Outros 430 pacientes, aqueles com diagnóstico de síndrome de Turner 272 (23,8%), de síndrome de Klinefelter 66 (5,7%) e de disgenesia gonadal pura 92 (8%), também foram excluídos. Portanto, foram incluídos 709 casos.
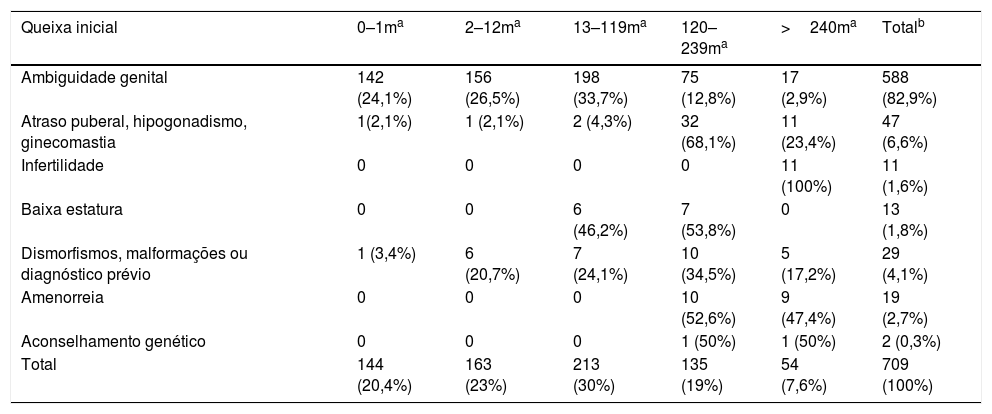
Na tabela 1 são apresentados os motivos de encaminhamento de acordo com o grupo etário. Houve predomínio de casos com ambiguidade genital, 588 casos (82,9%), 50,6% encaminhados nos primeiros 12 meses de idade. Dos 47 casos de atraso puberal, hipogonadismo ou ginecomastia, 91,5% (43) foram encaminhados após os 10 anos. Os pacientes encaminhados com menos de um ano foram diagnósticos precoces de hipogonadismo na minipuberdade, um com pan‐hipopituitarismo e o outro com microrquidia. Os 19 casos (2,7%) de amenorreia também foram encaminhados após os 10 anos. Os dois casos encaminhados para aconselhamento genético eram gestantes com histórico de gestação anterior de filhos com hiperplasia adrenal congênita.
Frequências da queixa inicial por grupo etário, em meses, dos 709 casos de DDS
| Queixa inicial | 0–1ma | 2–12ma | 13–119ma | 120–239ma | >240ma | Totalb |
|---|---|---|---|---|---|---|
| Ambiguidade genital | 142 (24,1%) | 156 (26,5%) | 198 (33,7%) | 75 (12,8%) | 17 (2,9%) | 588 (82,9%) |
| Atraso puberal, hipogonadismo, ginecomastia | 1(2,1%) | 1 (2,1%) | 2 (4,3%) | 32 (68,1%) | 11 (23,4%) | 47 (6,6%) |
| Infertilidade | 0 | 0 | 0 | 0 | 11 (100%) | 11 (1,6%) |
| Baixa estatura | 0 | 0 | 6 (46,2%) | 7 (53,8%) | 0 | 13 (1,8%) |
| Dismorfismos, malformações ou diagnóstico prévio | 1 (3,4%) | 6 (20,7%) | 7 (24,1%) | 10 (34,5%) | 5 (17,2%) | 29 (4,1%) |
| Amenorreia | 0 | 0 | 0 | 10 (52,6%) | 9 (47,4%) | 19 (2,7%) |
| Aconselhamento genético | 0 | 0 | 0 | 1 (50%) | 1 (50%) | 2 (0,3%) |
| Total | 144 (20,4%) | 163 (23%) | 213 (30%) | 135 (19%) | 54 (7,6%) | 709 (100%) |
Quanto aos cariótipos dos pacientes, três vezes mais indivíduos tinham cariótipo 46,XY (68,6%) do que 46,XX (22,4%). Anomalias de cromossomos sexuais caracterizadas por alterações estruturais ou numéricas, após descartadas as síndromes de Turner e Klinefelter, compreenderam 9% dos casos.
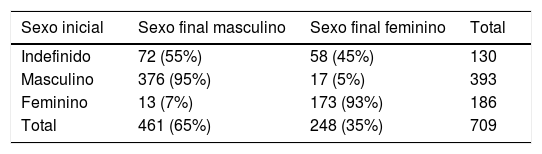
Na tabela 2 são mostradas as frequências dos sexos inicial e final e suas relações. Apenas 130 casos (18,3%) vieram com sexo social indefinido. Eles tinham idade até 12 meses, 99 (76,2%) encaminhados no primeiro mês de vida. Trinta casos (12%) necessitaram de mudança de sexo, 17 de masculino para feminino e 13 de feminino para masculino. No fim, foram 65% com sexo masculino e 35% com feminino.
Frequências do sexo inicial, ao encaminhamento e no fim, definidas após diagnóstico etiológico, dos 709 casos de DDS
| Sexo inicial | Sexo final masculino | Sexo final feminino | Total |
|---|---|---|---|
| Indefinido | 72 (55%) | 58 (45%) | 130 |
| Masculino | 376 (95%) | 17 (5%) | 393 |
| Feminino | 13 (7%) | 173 (93%) | 186 |
| Total | 461 (65%) | 248 (35%) | 709 |
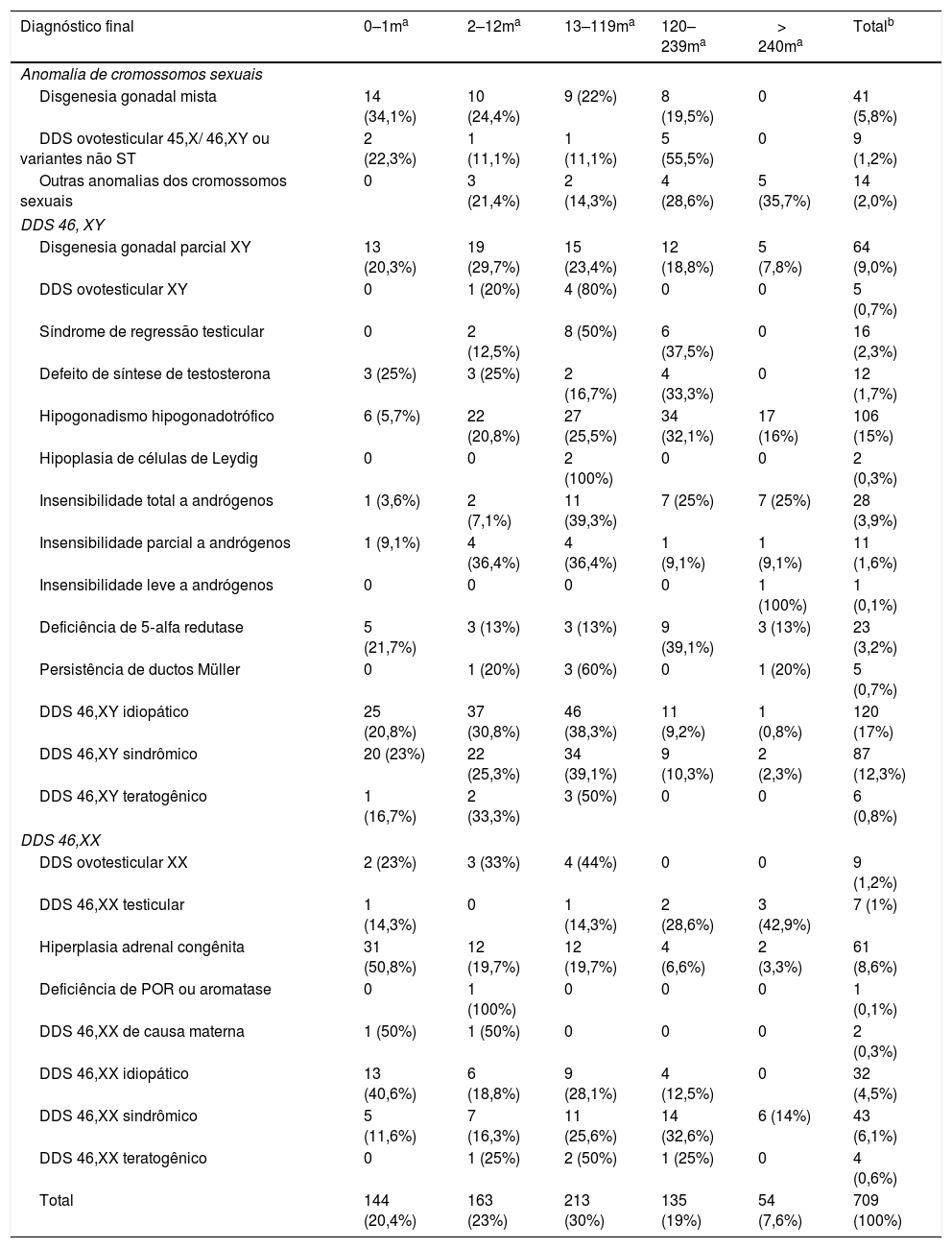
Em relação aos diagnósticos feitos, a tabela 3 mostra sua distribuição por grupo etário. Nota‐se que mais da metade, 486 pacientes (68,6%), tiveram diagnóstico de DDS 46,XY, 159 (22,4%) tiveram diagnóstico de DDS 46,XX e 64 (9%) apresentaram alterações de cromossomos sexuais. Entre os casos de anomalias de cromossomos sexuais, os diagnósticos de disgenesia gonadal mista predominaram antes dos 10 anos, com a mediana ao diagnóstico de quatro meses (IC 0?204), enquanto que os de DDS ovotesticular tiveram distribuição similar antes e após 10 anos, mediana de 120 meses (IC 0?228). Para as outras anomalias dos cromossomos sexuais o predomínio do diagnóstico foi após os 10 anos, mediana aos 210 meses (IC 2?408). Em relação aos DDS 46,XY, os casos de disgenesia gonadal parcial XY, síndrome de regressão testicular, DDS ovotesticular XY, idiopático e sindrômico tiveram o predomínio até os 10 anos, com as respectivas medianas e intervalos de confiança: 13,5 meses (0?324), 82 meses (2?216), 18 (6?300) e 14 meses (IC 0?420). O diagnóstico de insensibilidade total aos andrógenos foi maior após um ano, mediana 113 meses (IC 1?432), e os demais foram similares em todas as faixas etárias. Finalmente, em relação aos casos de DDS 46,XX, os pacientes com hiperplasia adrenal congênita tiveram predomínio de diagnóstico até um ano, a maioria dentro do primeiro mês de vida, mediana um mês (IC 0?432). Os demais diagnósticos tiveram distribuição semelhante nos grupos etários avaliados.
Frequências dos diferentes diagnósticos de DDS por grupo etário, em meses, dos 709 casos
| Diagnóstico final | 0–1ma | 2–12ma | 13–119ma | 120–239ma | > 240ma | Totalb |
|---|---|---|---|---|---|---|
| Anomalia de cromossomos sexuais | ||||||
| Disgenesia gonadal mista | 14 (34,1%) | 10 (24,4%) | 9 (22%) | 8 (19,5%) | 0 | 41 (5,8%) |
| DDS ovotesticular 45,X/ 46,XY ou variantes não ST | 2 (22,3%) | 1 (11,1%) | 1 (11,1%) | 5 (55,5%) | 0 | 9 (1,2%) |
| Outras anomalias dos cromossomos sexuais | 0 | 3 (21,4%) | 2 (14,3%) | 4 (28,6%) | 5 (35,7%) | 14 (2,0%) |
| DDS 46, XY | ||||||
| Disgenesia gonadal parcial XY | 13 (20,3%) | 19 (29,7%) | 15 (23,4%) | 12 (18,8%) | 5 (7,8%) | 64 (9,0%) |
| DDS ovotesticular XY | 0 | 1 (20%) | 4 (80%) | 0 | 0 | 5 (0,7%) |
| Síndrome de regressão testicular | 0 | 2 (12,5%) | 8 (50%) | 6 (37,5%) | 0 | 16 (2,3%) |
| Defeito de síntese de testosterona | 3 (25%) | 3 (25%) | 2 (16,7%) | 4 (33,3%) | 0 | 12 (1,7%) |
| Hipogonadismo hipogonadotrófico | 6 (5,7%) | 22 (20,8%) | 27 (25,5%) | 34 (32,1%) | 17 (16%) | 106 (15%) |
| Hipoplasia de células de Leydig | 0 | 0 | 2 (100%) | 0 | 0 | 2 (0,3%) |
| Insensibilidade total a andrógenos | 1 (3,6%) | 2 (7,1%) | 11 (39,3%) | 7 (25%) | 7 (25%) | 28 (3,9%) |
| Insensibilidade parcial a andrógenos | 1 (9,1%) | 4 (36,4%) | 4 (36,4%) | 1 (9,1%) | 1 (9,1%) | 11 (1,6%) |
| Insensibilidade leve a andrógenos | 0 | 0 | 0 | 0 | 1 (100%) | 1 (0,1%) |
| Deficiência de 5‐alfa redutase | 5 (21,7%) | 3 (13%) | 3 (13%) | 9 (39,1%) | 3 (13%) | 23 (3,2%) |
| Persistência de ductos Müller | 0 | 1 (20%) | 3 (60%) | 0 | 1 (20%) | 5 (0,7%) |
| DDS 46,XY idiopático | 25 (20,8%) | 37 (30,8%) | 46 (38,3%) | 11 (9,2%) | 1 (0,8%) | 120 (17%) |
| DDS 46,XY sindrômico | 20 (23%) | 22 (25,3%) | 34 (39,1%) | 9 (10,3%) | 2 (2,3%) | 87 (12,3%) |
| DDS 46,XY teratogênico | 1 (16,7%) | 2 (33,3%) | 3 (50%) | 0 | 0 | 6 (0,8%) |
| DDS 46,XX | ||||||
| DDS ovotesticular XX | 2 (23%) | 3 (33%) | 4 (44%) | 0 | 0 | 9 (1,2%) |
| DDS 46,XX testicular | 1 (14,3%) | 0 | 1 (14,3%) | 2 (28,6%) | 3 (42,9%) | 7 (1%) |
| Hiperplasia adrenal congênita | 31 (50,8%) | 12 (19,7%) | 12 (19,7%) | 4 (6,6%) | 2 (3,3%) | 61 (8,6%) |
| Deficiência de POR ou aromatase | 0 | 1 (100%) | 0 | 0 | 0 | 1 (0,1%) |
| DDS 46,XX de causa materna | 1 (50%) | 1 (50%) | 0 | 0 | 0 | 2 (0,3%) |
| DDS 46,XX idiopático | 13 (40,6%) | 6 (18,8%) | 9 (28,1%) | 4 (12,5%) | 0 | 32 (4,5%) |
| DDS 46,XX sindrômico | 5 (11,6%) | 7 (16,3%) | 11 (25,6%) | 14 (32,6%) | 6 (14%) | 43 (6,1%) |
| DDS 46,XX teratogênico | 0 | 1 (25%) | 2 (50%) | 1 (25%) | 0 | 4 (0,6%) |
| Total | 144 (20,4%) | 163 (23%) | 213 (30%) | 135 (19%) | 54 (7,6%) | 709 (100%) |
A mediana do grupo de DDS como um todo foi de 24 meses (0–696), enquanto que nos 591 casos de ambiguidade genital foi de 12 meses (0–696).
DiscussãoAté o presente momento, este estudo descreve a maior coorte mundial de pacientes de um único centro, abrange um período de 28 anos, com diagnóstico confirmado de um tipo de DDS, total de 1.139 casos. Isso é possível por se tratar de um centro de referência nacional em relação ao cuidado integral e de qualidade aos pacientes com DDS.
Casuísticas semelhantes verificadas em revisão de literatura são de países emergentes como o Brasil: África do Sul,10 346 pacientes avaliados em 20 anos; Tailândia,11 117 pacientes em 20 anos; Turquia,12 95 pacientes em três anos; Indonesia,13 286 pacientes em sete anos; India,14 194 pacientes em 20 anos. Um país desenvolvido, Finlândia,15 com 550 pacientes em 10 anos, e dois estudos multicêntricos europeus, o I‐DSD Registry,16 que está criando uma base de dados em DDS com 14 países e 649 pacientes, e o DSD Life,17 consórcio entre 16 centros hospitalares europeus, que avaliou a qualidade de vida de 1.040 pacientes maiores de maiores de 16 anos com DDS.
Embora seja possível argumentar que, ao levantar a suspeita de DDS em muitos pacientes, a assistência primária em saúde diminui o risco de perda de diagnóstico em alguns casos e, também, que alguns exames como o de cariótipo não sejam de fácil acesso fora de centros especializados, o fato de que mais de um terço dos pacientes encaminhados tenha sido caracterizado como erro de encaminhamento reforça a necessidade de programas de treinamento e de oferecimento de materiais didáticos e de apoio para que os médicos não especialistas, como neonatologistas, pediatras e clínicos, consigam encaminhar ao serviço terciário somente os pacientes que tenham indicação real, não onerem em demasia o sistema de saúde e não causem incertezas e preocupações desnecessárias às famílias.4,18
Optou‐se por excluir também da análise por faixas etárias os casos diagnosticados de síndrome de Turner, de Klinefelter e disgenesia gonadal pura, pois, embora estejam entre os diagnósticos mais comuns, tanto na casuística deste estudo quanto em outras coortes e em relação à prevalência mundial,5,8 são pacientes que normalmente não se apresentam com ambiguidade genital e não necessitam ser acompanhados em centros multiprofissionais especializados em DDS, dessa forma não agregam ao objetivo final de análise em relação ao processo de encaminhamento ao centro especializado. O estudo indiano foi o único que também optou por essa exclusão, pela mesma razão.14
Em todas as faixas etárias foram encaminhados pacientes com queixa inicial de ambiguidade genital. Entretanto, somente pacientes de até 12 meses não tinham registro em algum sexo, total de 130 pacientes, o que corresponde a menos da metade dos 298 pacientes com ambiguidade genital encaminhados nessa faixa etária.
Embora a não designação do sexo logo ao nascimento gere angústias na família e entraves burocráticos, já que esse ainda é um fator de forte peso social, o ideal é que ela seja feita após estudo adequado do caso e em discussão com parentes, leve em conta diagnóstico, possibilidades cirúrgicas, perspectiva de reposição hormonal e fertilidade, para que haja maior chance de que o sexo definido seja compatível com a identidade de gênero do indivíduo (que só será definida ao longo da vida) e que não seja necessário passar pelo processo de redesignação de sexo, seja na infância ou após.18
A maioria dos pacientes com sexo indefinido 76,2% foi encaminhada com até um mês. Aspecto positivo a ser destacado e que é de grande valia para as famílias, pela perspectiva de finalização mais precoce do processo de investigação e definição do sexo social. Entre os pacientes com sexo já definido ao encaminhamento, 55,4% eram do sexo masculino e 26,2% feminino.
Já em relação à designação de sexo final, 65% dos pacientes ficaram no sexo masculino e 35% no feminino. A redesignação de sexo foi feita somente em indivíduos com ambiguidade genital, ocorreu em 17 pacientes com registro inicial no sexo masculino (5%) e 13 inicialmente no feminino (7%). Na casuística turca, 39% dos pacientes tinham sexo indefinido ao encaminhamento; 23,1% masculino e 37,9% feminino. No fim, 44,3% ficaram como sexo definido masculino e 55,7% feminino.12
O principal motivo de encaminhamento neste estudo foi ambiguidade genital, em 82,9% dos pacientes. Embora a maioria deles tenha sido encaminhada até o primeiro ano, esse cenário ainda está aquém do ideal, no qual todos os casos com ambiguidade genital seriam detectados já na maternidade e o encaminhamento feito no momento de alta. Aqueles pacientes com ambiguidade genital que só foram encaminhados após os 20 anos, pior cenário possível, tiveram os mais diversos diagnósticos, em sua maioria aqueles que podem cursar com graus mais leves de ambiguidade genital e, portanto, identificados tardiamente. Entre eles, disgenesia gonadal parcial XY, hipogonadismo hipogonadotrófico, insensibilidade total aos andrógenos, deficiência de 5α‐redutase e persistência de ductos de Müller.
Conforme o esperado, a maioria dos pacientes com atraso puberal, hipogonadismo e/ou amenorreia foi encaminhada entre 10 e 19 anos. Entretanto, ainda houve encaminhamentos tardios após os 20 anos em 23,4% dos pacientes com atraso puberal/hipogonadismo e 47,4% daqueles com amenorreia, fato que atrasa o início da reposição com prejuízo do crescimento, redução da massa óssea e, consequentemente, compromete a qualidade de vida desses pacientes.
Outras casuísticas analisadas não separam os motivos de encaminhamento por faixa etária, somente sua frequência absoluta. No estudo tailandês,11 36,8% dos pacientes foram encaminhados por ambiguidade genital. Já no estudo turco, 24,2% foram encaminhados pela mesma causa,12 frequências semelhantes entre si, porém menores do que a frequência do presente estudo, de 82,9%. Atraso puberal foi motivo de encaminhamento em 8,4% dos pacientes no estudo turco, semelhante à da presente casuística (6,6%), enquanto amenorreia foi observada em 8,4%, maior do que deste estudo (2,2%).
Em relação aos grupos diagnósticos definidos pelo consenso de Chicago, no presente estudo 68,6% caracterizaram um DDS 46,XY, 22,4% DDS 46,XX e 9% DDS por anomalias dos cromossomos sexuais. As coortes de África do Sul,10 Indonésia,13 Índia14 e do I‐DSD Registry16 apresentaram proporções semelhantes, com mais da metade de DDS 46,XY, e as anomalias de cromossomos sexuais representaram no máximo 10% da coorte. A maior complexidade da diferenciação sexual masculina explica em parte a existência de maior número de casos de DDS com ambiguidade genital em indivíduos com cariótipo 46,XY.19 Já nos estudos tailandês,11 turco12 e no DSD Life,17 houve uma preponderância relativa das anomalias de cromossomos sexuais (53%, 27,4% e 54,3% respectivamente) em relação aos DDS 46, XY (17,1%, 47,3% e 21,4%). No estudo finlandês,15 por outro lado, houve maior número de anomalias de cromossomos sexuais (37,1%) em relação a casos de DDS 46,XX (9,6%). Razões para isso seria o fato de essas coortes não terem excluído os indivíduos com síndrome de Turner e Klinefelter e, no caso finlandês, pelo maior número de casos com diagnóstico pré‐natal das anomalias cromossômicas.
Por fim, quanto à frequência dos diagnósticos específicos e sua distribuição por idades, cabe destacar entre o grupo de DDS 46,XY que 3,9% tiveram diagnóstico de insensibilidade total aos andrógenos, com mediana ao diagnóstico de 9,4 anos. A proporção é similar ao grupo tailandês,11 com 5,1% de diagnósticos, porém nesse caso a mediana de idade foi de 3,3 anos, uma vez que muitos casos foram diagnosticados por presença de massa inguinal na infância. Já no estudo indiano,14 a frequência foi de 1,5%, com a mediana ao diagnóstico de 18 anos. Um estudo dinamarquês20 que avaliou somente casos de mulheres com cariótipo XY definiu a mediana de diagnóstico como 7,5 anos, também por ambiguidade genital ou histórico familiar.
Na disgenesia gonadal parcial XY, a frequência foi de 9% neste estudo, maior do que a sul africana10 e a do DSD Life,17 de 1,1% e 3,5%, respectivamente, porém semelhante às da Índia14 e Tailândia,11 de 5,6% e 6,0%. A mediana ao diagnóstico de nossa casuística foi de 1,1 ano, menor do que a tailandesa, de 4,3 anos.
Já em relação à síndrome de regressão testicular, a frequência de diagnóstico no presente estudo foi de 2,3%, com mediana de 6,8 anos, bastante aquém da finlandesa,15 de 4,9%, com mediana de 0,84 ano.
Para os casos de DDS 46,XX, é importante ressaltar o diagnóstico de hiperplasia adrenal congênita. A casuística de 8,6% é baixa em relação à mundial21 e à da maioria dos estudos, 26,5% tailandesa,11 16,8% turca,12 13,9% indonésia,13 26,8% indiana14 e 21,7% do DSD Life.17 Isso ocorre pois em nosso serviço existe outro ambulatório específico para hiperplasia adrenal, no qual se concentram os pacientes diagnosticados via triagem neonatal e as formas tardias que sejam encaminhadas. A mediana ao diagnóstico é de um mês, o que demonstra eficácia do sistema de triagem neonatal e é semelhante aos estudos da Índia e da Finlândia. A coorte sul‐africana10 tem prevalência semelhante à nossa (9%), porém isso se dá devido à ausência de diagnósticos pela falta da triagem neonatal no país. A finlandesa15 também é baixa, de 5,6%, porém isso é explicado pela proporcionalidade de DDS 46,XX nessa coorte ser baixa.
Quanto aos quadros de anomalias de cromossomos sexuais, a prevalência de disgenesia gonadal mista é aumentada em nosso estudo (5,8%) em relação ao sul‐africano (1,1%),10 ao turco (3,1%),12 ao indonésio (2,7%)13 e ao indiano (2,5%);14 similar à DSD Life,17 de 4,3%, e menor do que o tailandês,11 de 8,5%. A mediana ao diagnóstico no nosso caso foi baixa, de 0,33 ano, menor do que a indonésia e a indiana, de 9,5 e 10 anos, respectivamente.
De uma forma geral a mediana do grupo de DDS neste estudo foi de 24 meses, enquanto que para os 591 casos de ambiguidade genital foi de 12 meses. Apesar de relativamente baixa, ainda não está adequada, possivelmente reflete em parte o não reconhecimento de alguns casos logo ao nascimento, o desconhecimento do processo de investigação pelos pediatras que avaliam casos de DDS e a baixa disponibilidade de centros de investigação em um país como o nosso, de dimensões continentais.
Este estudo apresenta contribuições relevantes para a melhoria da atenção a pessoas com DDS, respaldado pela representatividade de sua casuística. A análise de diversos aspectos reforça a importância da educação continuada a profissionais responsáveis pelo atendimento inicial e o encaminhamento desses pacientes a serviços de referência, principalmente pediatras e neonatologistas, permite intervenções diagnóstico‐terapêuticas mais precoces e eficientes, de modo a reduzir complicações, tanto no âmbito físico como psicossocial desses indivíduos, propicia melhores perspectivas de adaptação e qualidade de vida às pessoas com DDS e suas famílias.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
Please cite this article as: El Beck MS, Germano CW, Barros BA, Andrade JG, Guaragna‐Filho G, Paula GB, et al. Por que os pediatras precisam conhecer os distúrbios da diferenciação do sexo: experiência de 709 casos de um serviço especializado. J Pediatr (Rio J). 2020;96:585–91.
Estudo vinculado à Universidade Estadual de Campinas (Unicamp), Faculdade de Ciências Médicas (FCM), Grupo Interdisciplinar de Estudos da Determinação e Diferenciação do Sexo (GIEDDS), Campinas, SP, Brasil.





