
To describe the demographic, clinical, laboratory and molecular characteristics of patients with lysosomal acid lipase deficiency.
MethodsA retrospective review of the medical records of children with the disease.
ResultsSeven children with lysosomal acid lipase deficiency (5 male; 2 female); 6 were mixed race, and 1 was black. The mean ages at the first onset of symptoms and at diagnosis were 5.0 years (4 months to 9 years) and 6.9 years (3–10 years), respectively. Symptom manifestations at onset were: 3 patients had abdominal pain, one had bone/joint pain due to rickets, and 1 had chronic diarrhea and respiratory insufficiency due to interstitial pneumonitis. One was asymptomatic, and clinical suspicion arose due to hepatomegaly. Six patients had hepatomegaly, and none had splenomegaly. Two patients were siblings. Enzymatic assay and molecular analysis confirmed the diagnoses. Genetic analysis revealed a rare pathogenic variant (p.L89P) in three patients, described only once in medical literature and never described in Brazil. None of those patients were related to each other. Lysosomal acid lipase deficiency was previously described as an autosomal recessive disease, but three patients were heterozygous and undoubtedly had the disease (low enzyme activity, suggestive lab findings and clinical symptoms).
ConclusionThis case series supports that lysosomal acid lipase deficiency can present with highly heterogeneous signs and symptoms among patients, but it should be considered in children presenting with gastrointestinal symptoms associated with dyslipidemia. We describe a rare variant in three non‐related patients that may suggest a Brazilian genotype for lysosomal acid lipase deficiency.
Descrever as características demográficas, clínicas, laboratoriais e moleculares de pacientes com deficiência de lipase ácida lisossomal.
MétodosAnálise retrospectiva dos prontuários médicos de crianças com a deficiência de lipase ácida lisossomal.
ResultadosSete crianças com deficiência de lipase ácida lisossomal (5M:2F); seis eram pardas e uma negra. As faixas etárias no início dos sintomas e no diagnóstico foram 5 anos (4 meses a 9 anos) e 6,9 anos (3 a 10 anos), respectivamente. As manifestações dos sintomas no início foram as que seguem: três pacientes apresentaram dor abdominal, um apresentou dor nos ossos/articulações devido a raquitismo e um apresentou diarreia crônica e insuficiência respiratória devido à pneumonite intersticial. Os outros não apresentaram sintomas e a suspeita clínica surgiu devido à hepatomegalia. Seis pacientes apresentaram hepatomegalia e um apresentou esplenomegalia. Dois pacientes eram irmãos. O ensaio enzimético e a análise molecular confirmaram os diagnósticos. A análise genética revelou uma variante patogênica rara (p.L89P) em três pacientes, descrita uma única vez na literatura médica e nunca descrita no Brasil. Nenhum desses pacientes tinha parentesco com os outros. A deficiência de lipase ácida lisossomal foi anteriormente descrita como uma doença recessiva autossômica, porém três pacientes eram heterozigotos e, sem dúvida, apresentaram a doença (atividade enzimática baixa, achados laboratoriais sugestivos e sintomas clínicos).
ConclusãoEsta casuística afirma que a deficiência de lipase ácida lisossomal pode se manifestar com sinais e sintomas altamente heterogêneos entre os pacientes, porém deve ser considerada em crianças que apresentam sintomas gastrointestinais associados à dislipidemia. Descrevemos uma variante rara em três pacientes não relacionados que pode sugerir um genótipo brasileiro para deficiência de lipase ácida lisossomal.
A deficiência de lipase ácida lisossomal (DLAL) é uma doença recessiva autossômica que pertence ao grupo de doenças de depósito lisossômico.1–3 O fenótipo da DLAL varia de acordo com a atividade da enzima LAL e o espectro clínico pode variar de doença de Wolman (DW), uma forma grave de início na infância, a doença do armazenamento de éster de colesterol (DAEC), uma doença com sintomas mais leves, porém às vezes com apresentação grave e prognóstico ruim.1–6 A baixa frequência da DLAL foi contestada, pois os estudos populacionais propõem que sua incidência pode ser maior do que a descrita anteriormente.2,3,7 Adicionalmente, o aumento da conscientização clínica resultou em mais diagnósticos, corroborou a ideia de que ela pode ser uma doença subdiagnosticada. Foram descritas muitas variantes no gene da lipase ácida lisossomal (LIPA) e novos estudos ainda escrevem novas mutações; atualmente, não foi encontrada uma correlação genótipo‐fenótipo.1,2,8,9
Neste estudo, analisamos sete pacientes com DLAL e acreditamos ser a maior série relatada na América do Sul.10
MétodosFizemos uma análise retrospectiva dos prontuários médicos dos pacientes com DLAL tratados em nossa clínica ambulatorial. Todos os dados com relação às características demográficas, clínicas, laboratoriais e moleculares foram recuperados e, então, analisados. Todos os pacientes e seus pais (ou responsáveis) estavam cientes da intenção de publicar seus prontuários, concordaram com isso e assinaram um consentimento informado, de acordo com as diretrizes do Comitê de Ética de nossa instituição. Este estudo foi aprovado pelo Comitê de Ética.
O Departamento de Laboratórios do Seattle Children's fez todos os testes da atividade da LAL e a análise molecular com o uso de amostra de sangue seco (DBS). Os testes da atividade da LAL foram feitos com o método descrito por Hamilton et al. em 2012: foi usado 4‐metilumbeliferil palmitato como substrato para a atividade da lipase total determinada; como sangue total para outras lipases, Lalistat 2, um inibidor de LAL, foi acrescentado à amostra para medir a atividade da lipase pancreática e, por fim, foi calculada a atividade da LAL. As unidades usadas foram pmol/hr/spt. Foi feito o sequenciamento de Sanger do gene LIPA em cada paciente com baixa atividade da LAL.11
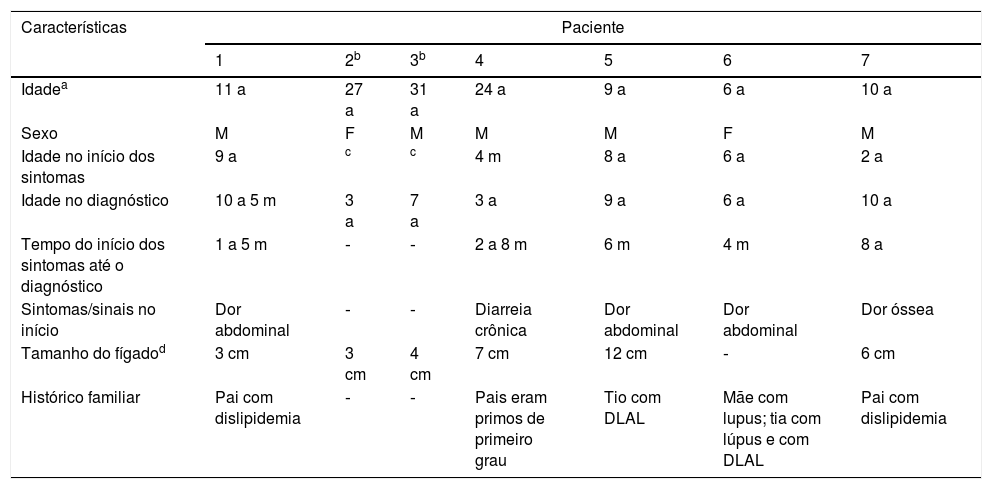
Resultados (descrições dos casos)Revisamos os prontuários médicos de sete crianças com DLAL (5M: 2 F); seis eram pardas e uma negra. As idades média e mediana no início dos sintomas foram cinco anos (quatro meses a nove anos) e seis anos e no diagnóstico foram 6,9 a (três a 10 anos) e 6,5 anos, respectivamente. O tempo entre o diagnóstico e o início dos sintomas variou amplamente e é mostrado na tabela 1, juntamente com outras características clínicas. Dois pacientes eram assintomáticos e a suspeita clínica surgiu devido à hepatomegalia. Seis pacientes apresentaram hepatomegalia e nenhum apresentou esplenomegalia nem desenvolveu icterícia. Dois pacientes irmãos apresentaram consanguinidade. Cinco dos sete pacientes apresentaram histórico familiar de dislipidemia. Após suspeita clínica e laboratorial em nossos pacientes, foi medida a atividade da LAL para confirmar DLAL.
Características clínicas
| Características | Paciente | ||||||
|---|---|---|---|---|---|---|---|
| 1 | 2b | 3b | 4 | 5 | 6 | 7 | |
| Idadea | 11 a | 27 a | 31 a | 24 a | 9 a | 6 a | 10 a |
| Sexo | M | F | M | M | M | F | M |
| Idade no início dos sintomas | 9 a | c | c | 4 m | 8 a | 6 a | 2 a |
| Idade no diagnóstico | 10 a 5 m | 3 a | 7 a | 3 a | 9 a | 6 a | 10 a |
| Tempo do início dos sintomas até o diagnóstico | 1 a 5 m | ‐ | ‐ | 2 a 8 m | 6 m | 4 m | 8 a |
| Sintomas/sinais no início | Dor abdominal | ‐ | ‐ | Diarreia crônica | Dor abdominal | Dor abdominal | Dor óssea |
| Tamanho do fígadod | 3 cm | 3 cm | 4 cm | 7 cm | 12 cm | ‐ | 6 cm |
| Histórico familiar | Pai com dislipidemia | ‐ | ‐ | Pais eram primos de primeiro grau | Tio com DLAL | Mãe com lupus; tia com lúpus e com DLAL | Pai com dislipidemia |
DLAL, deficiência de lipase ácida lisossomal.
Uma característica da DLAL é a heterogeneidade clínica dos sintomas na apresentação. É obrigatória uma descrição clínica detalhada dos casos.
O paciente 1 foi trazido a nossa instalação ambulatorial com dor abdominal, sem sinal alarmante, e tinha oito anos. Após avaliação contínua, observamos hepatomegalia e a endoscopia digestiva alta mostrou histiócitos azul marinho no exame histológico de biópsia da mucosa do duodeno. A medição da atividade da LAL confirmou a DLAL.
O paciente 3, durante uma consulta pediátrica de rotina aos seis anos, apresentou hepatomegalia. Os exames iniciais mostraram um aumento de 2‐3 vezes da alanina aminotransferase (ALT), baixo nível de lipoproteína de alta densidade (HDL) e nível elevado de lipoproteína de baixa densidade (LDL). A biópsia do fígado mostrou depósitos lipídicos nos histiócitos. Mais tarde, a atividade da LAL medida aos sete anos foi diagnóstico de DLAL. Sua irmã, a paciente 2, também apresentou enzimas hepáticas ligeiramente elevadas aos três anos, sem hepatomegalia, e foi diagnosticada ao mesmo tempo em que seu irmão com DLAL.
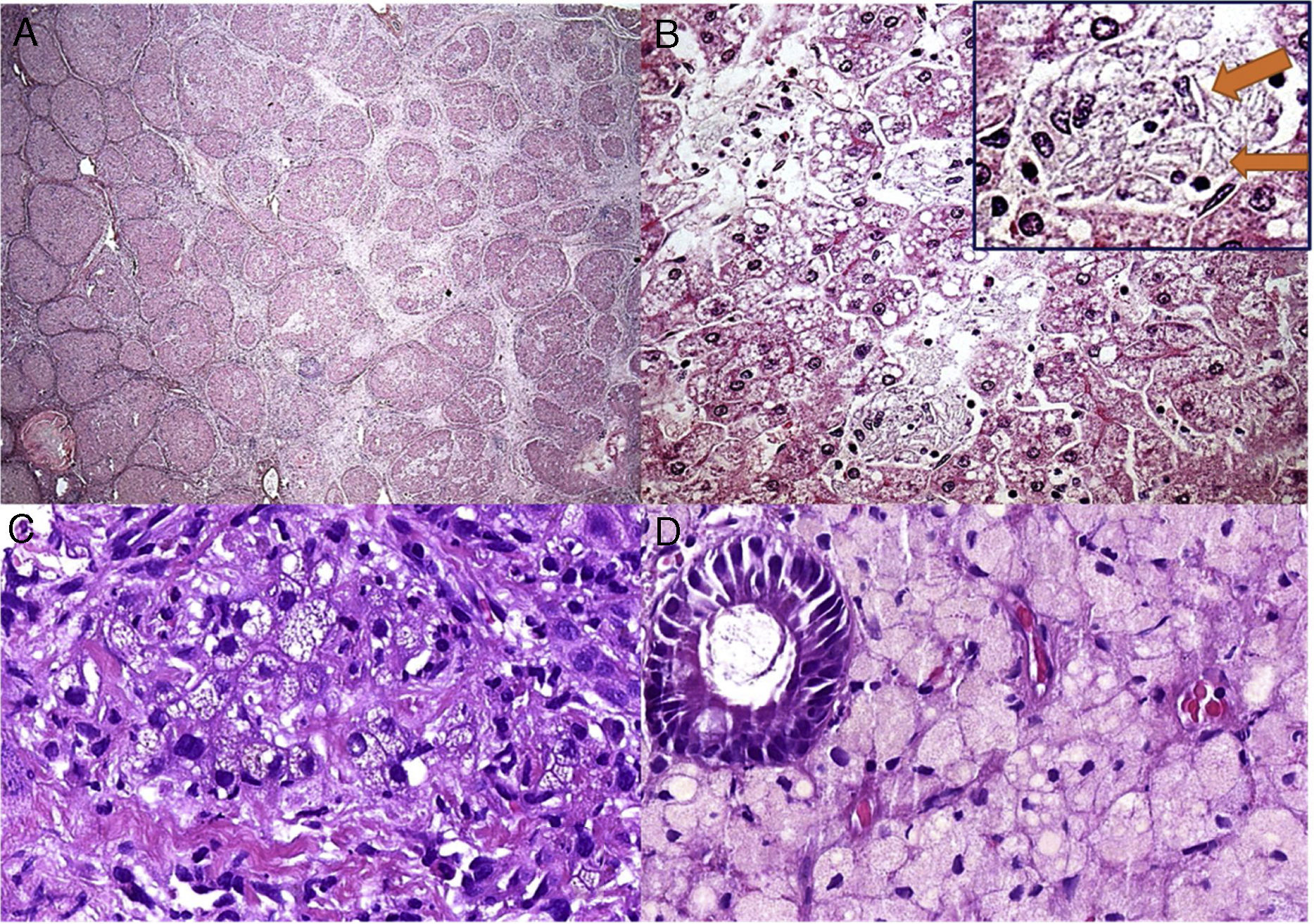
O paciente 4, que tinha quatro meses de idade, apresentou diarreia progressiva e intratável (primeira ocorrência em 1991), déficit de crescimento, hepatomegalia, dislipidemia e calcificações adrenais. Aos três anos, uma biópsia hepática revelou esteatose microvesicular significativa, fibrose septal e agregados de macrófagos com microvacuolização citoplasmática – achados sugestivos de Niemann‐Pick naquela época, pois também apresentou baixos níveis de esfingomielinase e níveis elevados de fosfatase ácida. Contudo, a microscopia eletrônica mostrou imagens negativas de cristais de colesterol e vacúolos lipídicos nos hepatócitos e histiócitos; assim, a DLAL foi favorecida por esse método. Sua diarreia crônica e doença hepática progressiva continuaram. Ao atingir seis anos, ele desenvolveu vários episódios de pieira. Os raios X e a tomografia de tórax mostraram sinais de pneumonite intersticial. A espirometria revelou um padrão restritivo. Aos 13 anos, desenvolveu complicações devido à insuficiência hepática com ascite e apresentou peritonite bacteriana espontânea duas vezes. Aos 18 anos, no início de 2010, foi submetido a uma cirurgia de transplante de fígado de um doador morto. Seu fígado explantado (fig. 1A e B) apresentou cirrose micronodular e características específicas da doença, inclusive algumas fendas em forma de agulha dentro das células Kupffer microvacuoladas. Antes do transplante de fígado, o paciente desenvolveu pequena esplenomegalia (2cm da borda costal esquerda) e a mesma apresentou redução de cerca de 50% após transplante de fígado. Após transplante do fígado, apresentou vários episódios de rejeição, que melhoraram com corticosteroides. Sua biópsia hepática em 2015 mostrou rejeição crônica com ductopenia (perda de 50%), fibrose portal e centrolobular, septos raros, perivenulite central e algumas células de Kupffer espumosas (geralmente são uma resposta a colestase nesse contexto). Contudo, sua biópsia hepática em 2017 mostrou muitos macrófagos xantomatosos no trato portal, fortemente sugestivo de recrudescimento da doença (fig. 1C), juntamente com fibrose em ponte marcada (F3), rejeição crônica e células sinusoidais espumosas raras. A atividade da enzima LAL medida em 2015 foi zero. O perfil lipídico do paciente melhorou para níveis normais de LDL e triglicerídeos (TG) após transplante de fígado, porém os baixos níveis de HDL continuaram. Além disso, uma biópsia duodenal feita em 2016 mostrou inúmeros macrófagos xantomatosos na lâmina própria (fig. 1D).
; B – Essa área (do explante do fígado) descreve esteatose microvesicular e mediovesicular em muitos hepatócitos e grupos de células de Kupffer xantomatosas em sinusoides (H&E, 200X), além de raras fendas em forma de agulha (setas) dentro das células de Kupffer na inserção; C – Muitos macrófagos no trato portal mostram citoplasma xantomatoso no enxerto hepático após sete anos de transplante de fígado (H&E, 400X); D – A lâmina própria é infiltrada intensamente por grandes células espumosas carregadas de lipídios nessa biópsia duodenal (H&E, 400X).")
Alguns aspectos histológicos do fígado e duodeno no paciente n° 4. A – Cirrose micronodular avançada observada no explante do fígado (H&E, 25X); B – Essa área (do explante do fígado) descreve esteatose microvesicular e mediovesicular em muitos hepatócitos e grupos de células de Kupffer xantomatosas em sinusoides (H&E, 200X), além de raras fendas em forma de agulha (setas) dentro das células de Kupffer na inserção; C – Muitos macrófagos no trato portal mostram citoplasma xantomatoso no enxerto hepático após sete anos de transplante de fígado (H&E, 400X); D – A lâmina própria é infiltrada intensamente por grandes células espumosas carregadas de lipídios nessa biópsia duodenal (H&E, 400X).
O paciente 5, com oito anos, apresentou dor abdominal crônica juntamente com hepatomegalia e aumento da aspartato aminotransferase (AST) e ALT, juntamente com baixo HDL e alto LDL. Após investigação inicial inconclusiva, uma biópsia de fígado revelou achados sugestivos de doença de Niemann‐Pick. Como o curso clínico não foi sugestivo de Niemann‐Pick, a medição da atividade da LAL diagnosticou DLAL. Sua tia também foi diagnosticada anteriormente com doença de Niemann‐Pick e, mais tarde, com DLAL.
O paciente 6 apresentou dor abdominal crônica por volta dos seis anos. A avaliação laboratorial inicial mostrou ligeiro aumento na AST e ALT, HDL baixa e LDL alta e esteatose no ultrassom do fígado. Ele não estava acima do peso e não havia consumido altas doses de frutose, sinais sugestivos de doença hepática gordurosa não alcoólica (DHGNA) ou esteatose hepática. Portanto, medimos a atividade da LAL e confirmamos DLAL.
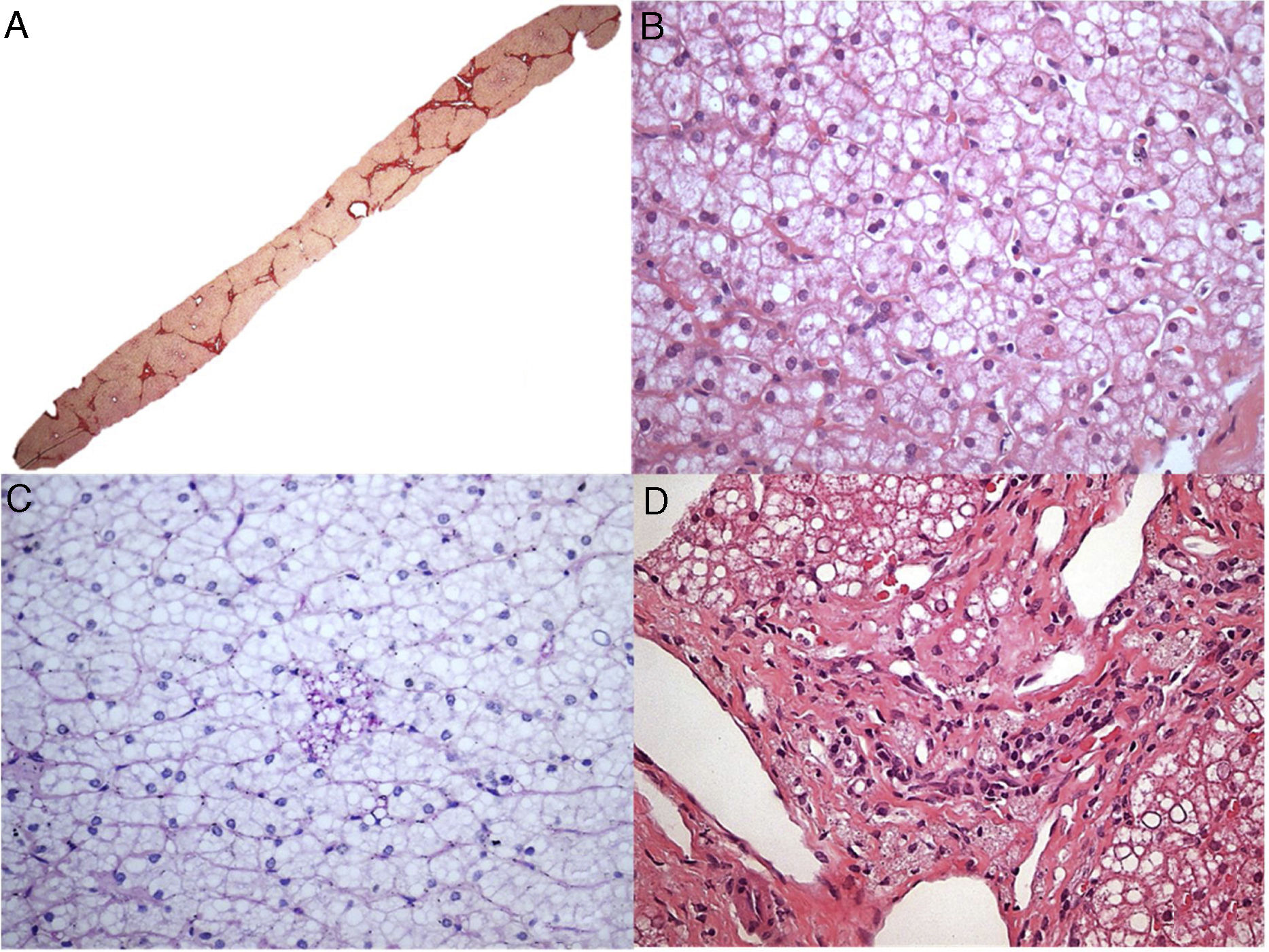
O paciente 7, um menino de dois anos, foi internado em nosso hospital devido à dificuldade de andar. Após exame físico inicial, detectamos hepatomegalia. A análise laboratorial indicou claramente raquitismo. Aos três anos, o paciente foi transferido para nossa clínica de hepatologia para investigar hepatomegalia. Uma biópsia hepática, feita quando o paciente tinha seis anos, sugeriu doenças metabólicas, principalmente glicogenose. Contudo, o paciente nunca desenvolveu hipoglicemia. Em 2016, após medir a atividade da LAL, diagnosticamos DLAL. Após um tempo, o patologista reavaliou as lâminas da biópsia hepática. Os achados histopatológicos (fig. 2) das poucas células de Kupffer espumosas e alguns macrófagos do trato portal fracamente positivos para ácido periódico de Schiff (PAS) após digestão diastásica, a ausência das células hepáticas inchadas características que se assemelham a células vegetais, sem mencionar a presença de esteatose grau 3, são sinais de diagnóstico de DLAL (além de outros diagnósticos diferenciais, como mucopolissacaridoses). Vale mencionar que esse caso mostrou somente mudanças sutis nas células de Kupffer, tornou o diagnóstico mais desafiador, possivelmente relacionado à variabilidade de amostragem das biópsias de fígado.
; B – Esteatose mediovesicular (variante de pequenas gotículas da esteatose macrovesicular) e hepatócitos com citoplasma pálido (H&E, 400X); C – Somente raras células de Kupffer microvacuoladas foram identificadas, que foram ligeiramente coradas com PAS (PAS após digestão diastásica, 400X); D – Alguns macrófagos com citoplasma espumoso e corado foram detectados nos tratos portais (H&E, 400X).")
Aspectos histológicos da biópsia de fígado no paciente 7. A – Fibrose em ponte marcada com nódulos ocasionais (coloração com Picrosirius Red, 25X); B – Esteatose mediovesicular (variante de pequenas gotículas da esteatose macrovesicular) e hepatócitos com citoplasma pálido (H&E, 400X); C – Somente raras células de Kupffer microvacuoladas foram identificadas, que foram ligeiramente coradas com PAS (PAS após digestão diastásica, 400X); D – Alguns macrófagos com citoplasma espumoso e corado foram detectados nos tratos portais (H&E, 400X).
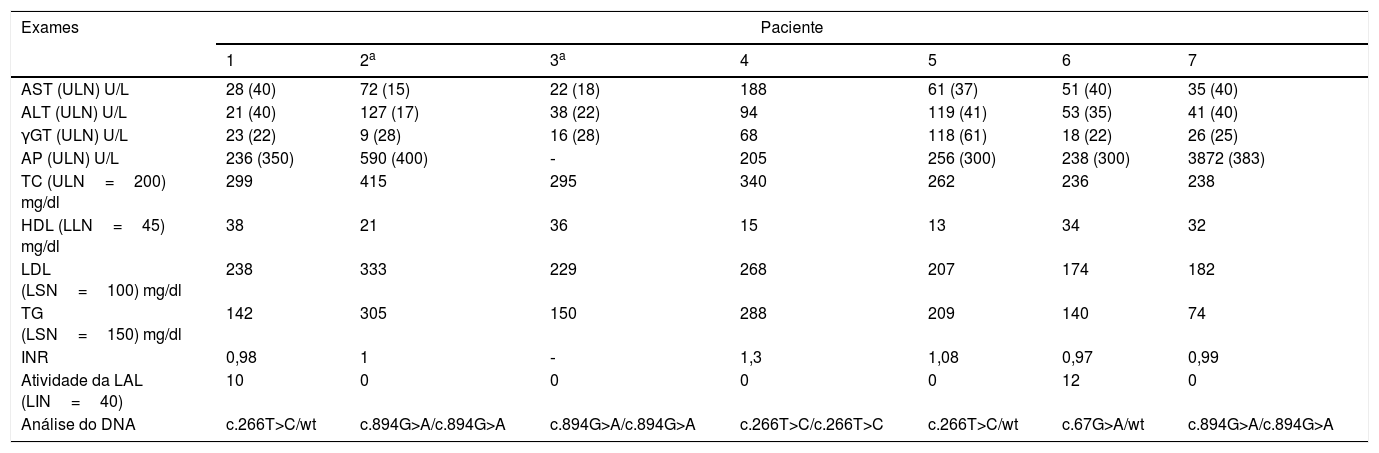
A tabela 2 resume as características laboratoriais de nossos pacientes. A análise genética revelou uma rara variante p.L89P (c.266T>C) em três pacientes, algo nunca descrito no Brasil.12 Nenhum desses pacientes estava relacionado com o outro. Um era homozigoto para p.L89P e os outros eram heterozigotos. As variantes c.894G>A, uma variante no local de porte na extremidade 3’ do éxon 8, e c.67G>A encontrada em nossos pacientes, já foram comumente descritas.
Trabalho laboratorial no início dos sintomas e análise genética
| Exames | Paciente | ||||||
|---|---|---|---|---|---|---|---|
| 1 | 2a | 3a | 4 | 5 | 6 | 7 | |
| AST (ULN) U/L | 28 (40) | 72 (15) | 22 (18) | 188 | 61 (37) | 51 (40) | 35 (40) |
| ALT (ULN) U/L | 21 (40) | 127 (17) | 38 (22) | 94 | 119 (41) | 53 (35) | 41 (40) |
| γGT (ULN) U/L | 23 (22) | 9 (28) | 16 (28) | 68 | 118 (61) | 18 (22) | 26 (25) |
| AP (ULN) U/L | 236 (350) | 590 (400) | ‐ | 205 | 256 (300) | 238 (300) | 3872 (383) |
| TC (ULN=200) mg/dl | 299 | 415 | 295 | 340 | 262 | 236 | 238 |
| HDL (LLN=45) mg/dl | 38 | 21 | 36 | 15 | 13 | 34 | 32 |
| LDL (LSN=100) mg/dl | 238 | 333 | 229 | 268 | 207 | 174 | 182 |
| TG (LSN=150) mg/dl | 142 | 305 | 150 | 288 | 209 | 140 | 74 |
| INR | 0,98 | 1 | ‐ | 1,3 | 1,08 | 0,97 | 0,99 |
| Atividade da LAL (LIN=40) | 10 | 0 | 0 | 0 | 0 | 12 | 0 |
| Análise do DNA | c.266T>C/wt | c.894G>A/c.894G>A | c.894G>A/c.894G>A | c.266T>C/c.266T>C | c.266T>C/wt | c.67G>A/wt | c.894G>A/c.894G>A |
γGT, gama glutamil transferase; ALT, alanina aminotransferase; AST, aspartato aminotransferase; CT, colesterol total; FA, fosfatase alcalina; HDL, lipoproteína de alta densidade; LDL, lipoproteína de baixa densidade; LIN, limite inferior da normalidade; LSN, limite superior da normalidade; RNI, razão normalizada internacional; TG, triglicerídeos; ts, gene tipo selvagem.
Relatamos sete pacientes com DLAL, a maior casuística já descrita na América do Sul. Os diagnósticos dos pacientes foram confirmados após alta suspeita clínica que levou à medicação da atividade da LAL, que foi invariavelmente baixa. Nossa conscientização clínica da DLAL melhorou ao longo dos anos e isso explica os intervalos de tempo variáveis entre o início dos sintomas e o diagnóstico. Os três pacientes com relação aos quais levamos mais de um ano para chegar ao diagnóstico são aqueles que apresentaram sintomas atípicos no acometimento.
O paciente 4 apresentou sintomas severos que se assemelham à doença de Wolman, porém também apresentou pneumonite intersticial, baixa esfingomielinase e altos níveis de fosfatase ácida, que são variáveis de confusão, pois esses achados são sugestivos de doença de Niemann‐Pick.
O paciente 7 apresentou hepatomegalia e a biópsia hepática inicial sugeriu uma doença de armazenamento, como glicogenose, pois alguns achados (por exemplo, células de Kupffer espumosas) não foram muito visíveis na amostra do tecido. A reavaliação das lâminas do fígado, após considerar as novas informações de DLAL, foi compatível com a doença.
Esses 2 casos mencionados (dos pacientes 4 e 7) acrescentaram a DLAL como um possível diagnóstico diferenciado de glicogenose e Niemann‐Pick tipo B. A doença de Gaucher, Niemann‐Pick tipo C e NAFLD já foram descritas como diagnósticos diferenciais de DLAL.13,14 Os diagnósticos diferenciais de DLAL são amplos e geralmente sobrepõem outras doenças crônicas. Consequentemente, uma razão pela qual a prevalência real da DLAL ser desconhecida é o fato de ela ser uma doença subdiagnosticada.2,3,7–9,15,16 Deve haver alta suspeita clínica de DLAL em pacientes com hepatomegalia não explicada, altos níveis de LDL e triglicerídeos, baixos níveis de HDL e enzimas hepáticas levemente elevadas.1,2,8 Seis de nossos pacientes apresentaram hepatomegalia, todos apresentaram um padrão de colesterol típico e cinco apresentaram enzimas hepáticas levemente elevadas. Esses resultados mostram que há um padrão de achados para diagnosticar DLAL, mesmo em casos atípicos.
O LIPA, o gene que codifica a LAL, está localizado no cromossomo 10q23.31. Sabe‐se que muitas variantes nesse gene estão associadas à DLAL. A variante c.894+1G>A (E8SJM+1) é a mais comum e é responsável por mais da metade dos casos publicados; três de nossos pacientes apresentaram essa variante.1,2 A outra variante, c.67G>A, já foi relatada três vezes no site ClinVar.17 Curiosamente, descrevemos neste trabalho uma variante rara, a c.266T>C (p.L89P), que foi encontrada em três pacientes. Essa variante patogênica foi descrita somente uma vez em uma apresentação em pôster na Reunião de 2014da Sociedade Americana de Genética Humana.12 Propusemos um possível genótipo brasileiro para a DLAL, pois nenhum dos pacientes com essa variante estava relacionado a outro. O paciente homozigoto para a última variante mostrou a apresentação mais grave com envolvimento pulmonar, diarreia crônica e doença hepática progressiva, necessitou de transplante de fígado (paciente 4).
A DLAL é classicamente definida como uma doença recessiva autossômica. Contudo, os pacientes 1, 5 e 6 apresentaram um alelo mutante e um alelo tipo selvagem. Todos esses pacientes apresentaram sinais laboratoriais e clínicos sugestivos de DLAL (hepatomegalia, baixa HDL, alta LDL e baixa atividade da LAL) e foram excluídas outras doenças prováveis. A doença de Niemann‐Pick foi o principal diagnóstico de confusão para o paciente 5, pois ele tinha uma tia com a doença de Niemann‐Pick. Contudo, após nosso diagnóstico, investigamos sua tia e pudemos diagnosticá‐la com DLAL. DHGNA e intolerância à frutose foram outros diagnósticos excluídos, pois elas podem surgir com hepatomegalia, esteatose hepática e especificamente DHGNA com dislipidemia. Outra hipótese é que esses pacientes podem ter mutações intrônicas que não foram reconhecidas por meio do sequenciamento de Sanger ou que a redução da atividade da LAL em alguns pacientes heterozigotos pode causar os sintomas. Pullinger et al. já descreveram os pacientes heterozigotos para as variantes patogênicas do gene LIPA que apresentaram sintomas e os dados laboratoriais sugestivos de DLAL.2,18
O tratamento e o prognóstico da DLAL mudaram drasticamente nos últimos anos após a criação do sebelipase alfa (Alexion, Kanuma®, Kanuma™, WA, EUA), uma terapia de reposição enzimática (TRE) que mostrou melhorias no dano hepático e nas anomalias lipídicas.19–22 O transplante de fígado para DLAL é outro possível tratamento para casos graves e foi relatado somente 20 vezes na literatura médica, porém poucos trabalhos relatam acompanhamento de longo prazo desses pacientes.2 Bernstein et al. publicaram dois casos de pacientes com DLAL submetidos a transplante hepático e, nos dois, a doença DLAL multissistêmica reapareceu após o transplante. Eles também revisaram a literatura de 18 casos de pacientes com DLAL submetidos a transplante de fígado e descreveram a progressão multissistêmica da DLAL em 11 pacientes (66%) e óbito em seis pacientes (33%).23 Em nosso estudo, o paciente 4 foi submetido a transplante de fígado e, após cinco anos de acompanhamento, apresentou rejeição crônica e manteve baixos níveis de HDL. Curiosamente, esse paciente ainda tinha deposição de colesterol nos macrófagos e mostrou histiócitos xantomatosos na biópsia de duodeno em 2016 e, mais tarde, desenvolveu macrófagos espumosos no trato portal na biópsia de fígado após sete anos de transplante de fígado. Portanto, o transplante de fígado não é a cura para essa doença, pois os macrófagos continuam a ser prejudicados, causam sintomas, pois outros órgãos, como a medula óssea e o intestino delgado, ainda conterão os macrófagos carregados de lipídios. Por outro lado, acredita‐se que o transplante de medula óssea (TMO) corrige o defeito da enzima e, portanto, se feito no início do curso da doença, pode salvar a vida dos pacientes com a doença de Wolman. Foram relatados alguns TMOs bem‐sucedidos na DLAL; contudo, a maior parte dos casos tem um prognóstico ruim e a TER imediata é a terapia de escolha.24–26
Em conclusão, relatamos a maior casuística de pacientes com DLAL na América do Sul e descrevemos um genótipo raro, possivelmente um genótipo brasileiro. Enfatizamos que a DLAL pode apresentar sinais e sintomas heterogêneos, porém ela deve ser incluída como um diagnóstico diferenciado de hepatomegalia, enzimas hepáticas elevadas e dislipidemia.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
Como citar este artigo: Benevides GN, Miura IK, Person NC, Pugliese RP, Danesi VL, Lima FR, et al. Lysosomal acid lipase deficiency in Brazilian children: a case series. J Pediatr (Rio J). 2019;95:552–8.





