

To verify genetic determinants associated with stroke in children with sickle cell disease (SCD).
MethodsProspective cohort with 110 children submitted to neonatal screening by the Neonatal Screening Program, between 1998 and 2007, with SCD diagnosis, followed at a regional reference public service for hemoglobinopathies. The analyzed variables were type of hemoglobinopathy, gender, coexistence with alpha thalassemia (α‐thal), haplotypes of the beta globin chain cluster, and stroke. The final analysis was conducted with 66 children with sickle cell anemia (SCA), using the chi‐squared test in the program SPSS® version 14.0.
ResultsAmong children with SCD, 60% had SCA. The prevalence of coexistence with α‐thal was 30.3% and the Bantu haplotype (CAR) was identified in 89.2%. The incidence of stroke was significantly higher in those with SCA (27.3% vs. 2.3%; p=0.001) and males (24.1% vs. 9.6%; p=0.044). The presence of α‐thal (p=0.196), the CAR haplotype (p=0.543), and socioeconomic factors were not statistically significant in association with the occurrence of stroke.
ConclusionThere is a high incidence of stroke in male children and in children with SCA. Coexistence with α‐thal and haplotypes of the beta globin chain cluster did not show any significant association with stroke. The heterogeneity between previously evaluated populations, the non‐reproducibility between studies, and the need to identify factors associated with stroke in patients with SCA indicate the necessity of conducting further research to demonstrate the relevance of genetic factors in stroke related to SCD.
Verificar fatores genéticos associados ao acidente vascular encefálico (AVE) em crianças com doença falciforme (DF).
MétodosCoorte prospectiva de 110 crianças submetidas à triagem neonatal pelo Programa de Triagem Neonatal, entre 1998‐2007, com o diagnóstico de DF, atendidas em serviço público regional de referência em hemoglobinopatias. As variáveis analisadas foram: tipo de hemoglobinopatia, sexo, coexistência da alfa‐Talassemia (α‐Tal), haplótipos do cluster da cadeia beta globina e AVE. A análise estatística final foi feita com 66 crianças com anemia falciforme, por meio do teste do qui‐quadrado no programa SPSS® 14.0.
ResultadosEntre as crianças com DF, 60% eram portadoras de anemia falciforme. A prevalência da coexistência com a α‐Tal foi de 30,3% e o haplótipo Bantu (CAR) foi identificado em 89,2%. A incidência de AVE foi significativamente maior nas crianças com AF (27,3% versus 2,3%; p = 0,001) e no sexo masculino (24,1% versus 9,6%; p = 0,044. A presença da α‐Tal (p = 0,196), do haplótipo CAR (p = 0,543) e de fatores socioeconômicos não foi significantemente associada à ocorrência de AVE.
ConclusãoO AVE apresenta alta incidência em crianças com AF e em crianças do sexo masculino. Coexistência de α‐Tal ou de haplótipos do cluster da betaglobina não apresentaram associação significante com AVE. A heterogeneticidade entre as populações previamente avaliadas e a não reprodutibilidade entre estudos indicam a necessidade de novas pesquisas para verificar o papel desses fatores genéticos no AVE em crianças com DF.
A doença falciforme (DF) é a doença hereditária monogênica mais comum no Brasil, ocorre predominantemente entre negros. O termo DF inclui anemia falciforme (AF) e condições patológicas em que o gene da hemoglobina S está associado a outras hemoglobinopatias hereditárias, tais como SC, S/beta0 e S/beta+ talassemia (S/b) e SD Punjab, entre outras.1 A AF, causada por uma única mutação no gene da β‐globina, produz uma diversidade de expressões fenotípicas nos pacientes acometidos.2,3 A AF é a forma mais grave de apresentação da DF e para que a doença se manifeste é necessário que ocorra homozigose dos alelos βS no gene responsável pela síntese da cadeia β da hemoglobina, o que determina a formação da hemoglobina S (HbSS). A HbSS em condições como baixa oxigenação, acidose metabólica ou desidratação se polimeriza e altera a estrutura da hemácia de forma irreversível, o que determinando oxigenação ineficiente, reação inflamatória endotelial e toda a complexa fisiopatologia da doença.3,4 O processo de polimerização leva à oclusão vascular, que pode desencadear crises álgicas, acidente vascular encefálico (AVE), síndrome torácica aguda, sequestro esplênico e priapismo, entre outras manifestações.
No Brasil os estudos mostram que nascem 700 a 1.000 crianças por/ano com DF.1,4–6 Isso faz com que essa patologia seja um problema de saúde pública. O Estado de Minas Gerais é pioneiro no Brasil no diagnóstico precoce das DF, com a introdução do Programa de Triagem Neonatal (PTN‐MG) em março de 1998. O PTN‐MG é coordenado pelo Núcleo de Ações e Pesquisa em Apoio Diagnóstico (Nupad) da Universidade Federal de Minas Gerais (UFMG), que encaminha os recém‐nascidos diagnosticados com DF para acompanhamento na Fundação Hemominas. Entre 1998 a 2007, foram triados em Minas 2.549.097 crianças, das quais 188.916 nasceram nas 39 cidades de abrangência do serviço público regional de referência em hemoglobinopatias – onde este estudo foi feito –, o que representa 7,41% do montante do estado. A cobertura do PTN‐MG em 2007 foi de 88,57%, percentagem superior em 9,65% à média nacional (Ministério da Saúde, Programa Nacional de Triagem Neonatal, 2009).
O AVE é uma das complicações mais graves da DF e é responsável por 20% da mortalidade dos pacientes.2,7 De acordo com o Grupo Cooperativo de Estudo em DF, a incidência global do primeiro AVE foi de 0,08 evento agudo/100 pacientes/ano em menores de dois anos; 0,75 em pacientes entre dois e cinco anos; 0,55 entre seis e nove anos; 0,30 entre 10 e 19 anos; e 0,45 entre 20 e 29 anos. Entre as DF, a incidência de acidentes vasculares cerebrais é de 0,61 para pacientes com Hb SS, 0,17 para Hb SC e 0,11 para Sbeta Talassemia.1,2,7
Atualmente o Doppler transcraniano (DTC), método ultrassonográfico não invasivo que mensura as velocidades e a alteração do fluxo dos vasos intracerebrais, é considerado uma ferramenta sensível para a identificação de risco para AVE isquêmico.1
A relação entre AVE, coexistência da alfa‐Talassemia(delα‐3,7) (α‐Tal) e os haplótipos do cluster da betaglobina βS é variável na literatura. Pesquisas publicadas com dados do Rio de Janeiro,4,8 Minas Gerais9 e São Paulo4,9 apresentam resultados controversos. Alguns estudos relatam que a coexistência da α‐Tal contribuiria para reduzir o risco de AVE,9–12 ao passo que outros não evidenciaram tal relação.6,13
Esses estudos identificaram características próprias quanto à frequência dos diferentes haplótipos, com grande prevalência de indivíduos homozigotos para o haplótipo CAR e Benin, o que reflete a origem do fluxo de escravos africanos recebidos na época do Brasil colônia.4,9,14 Na Jamaica e nos Estados Unidos o haplótipo Benin é muito mais frequente do que na República Centro‐Africana (RCA). Tais diferenças genéticas tornam inadequadas generalizações dos resultados entre as regiões.
O objetivo deste estudo foi verificar fatores genéticos associados ao risco de AVE em crianças com DF.
MétodoEntre 1998 e 2007, 188.916 crianças nascidas na Zona da Mata Mineira e Vertentes foram submetidas à triagem neonatal para DF por meio do PTN‐MG. Nesse período foram encaminhadas 135 crianças com DF para a instituição pública de referência em hemoglobinopatias. Desse total, nove foram a óbito antes do início do projeto, seis com DF tipo SD e S‐ foram excluídas e em dez houve perda de acompanhamento. Restaram 110 crianças, que constituem a população deste estudo.
O diagnóstico confirmatório de DF foi feito por meio de eletroforese de hemoglobina em pH alcalino aos seis e 12 meses, dosagem de hemoglobina A2 por cromatografia, imunodifusão radial para HbFetal e análise molecular do Códon 6da betaglobina. Foram consideradas para inclusão no estudo crianças com AF, SC, S/b0 e S/b+.
As informações clínicas e laboratoriais foram extraídas dos prontuários médicos, da data de cadastro na instituição até 31 de dezembro de 2013 (término do acompanhamento da coorte), o que permitiu um acompanhamento de no mínimo cinco anos dos sujeitos da pesquisa. As características socioeconômicas foram obtidas por meio da aplicação do questionário do Instituto Nacional de Estudos e Pesquisas Educacionais Anísio Teixeira (Inep), acrescido de questões sobre a escolaridade do cuidador e a renda familiar. Em novembro de 2010, foi iniciada a identificação dos haplótipos e a pesquisa para α‐Tal, após aprovação pela agência de fomento por meio do Programa Para a Saúde do SUS (PPSUS)/Fundação de Amparo à Pesquisa do Estado de Minas Gerais (Fapemig).
A variável de desfecho foi a presença de AVE (sim ou não). O diagnóstico de AVE foi feito clinicamente (AVE isquêmico ou ataque isquêmico transitório) ou por meio de exames complementares tais como doppler transcraniano (DTC) e angiorressonância magnética dos vasos cerebrais (AngioRM).
O rastreamento para a presença de AVE por meio do DTC tem sido oferecido anualmente a todas as crianças e adolescentes em acompanhamento de DF, entre dois e 16 anos, conforme preconizado pelo Protocolo Brasileiro, desde 2007 em Belo Horizonte e a partir de 2012 na unidade regional deste estudo. O exame é feito em todos pacientes com DF, apesar de não haver parâmetros de velocidades definidos para DF tipo SC e S/beta+.1 A determinação da velocidade do fluxo sanguíneo cerebral nas grandes artérias do Polígono de Willis foi baseada nos critérios do estudo Stroke Prevention Trial in Sickle Cell Anemia (Stop).1
Os resultados do DTC foram estratificados de acordo com a classificação proposta pelo estudo Stop, acrescentou‐se a recomendação de se incluir a velocidade máxima média (VMM) em uma das artérias cerebrais anteriores ≥ 170cm/s e também representou risco elevado de desenvolvimento de AVC isquêmico.15 As principais artérias insonadas foram: cerebrais médias e suas bifurcações e carótidas internas distais. A confirmação de teste anormal (risco alto) foi feita por dupla repetição do exame com intervalo de uma a quatro semanas. Crianças confirmadas como de alto risco para desenvolvimento de AVC isquêmico foram encaminhadas para tratamento preventivo primário do evento com regime de hipertransfusão. No momento dos DTC não havia crianças em terapia transfusional de troca nem em tratamento com hidroxiureia.
Os fatores genéticos considerados na análise foram:
- 1.
Tipo de DF (AF ou as demais: SC, S/b0 e S/b+ talassemia) com diagnóstico confirmado por metodologia descrita acima;
- 2.
Sexo (masculino ou feminino);
- 3.
Presença ou ausência da mutação para o gene da α‐Tal;
- 4.
Identificação do haplótipo (CAR ou não CAR).
Para a determinação da mutação da deleção para α‐Tal e haplótipos foram coletados 5ml de sangue total em tubos com etilenodiaminotetracético (EDTA) durante a rotina laboratorial de acompanhamento de DF. A extração e a quantificação do DNA genômico foram feitas com o kit comercial QIAamp DNA Blood Mini Kit (QIAGEN®, EUA) e Invitrogen® (Invitrogen Corporation®, EUA), com as enzimas de restrição (BioLabs Inc®, EUA) de acordo com instruções do fabricante. As análises foram feitas no laboratório de pesquisa da instituição.
Os haplótipos foram identificados com a técnica de reação em cadeia da polimerase (PCR) e a análise de polimorfismo de tamanho dos fragmentos de restrição (RFLP), de acordo com protocolo de Sutton.16 Para as reações de PCR e pesquisa de mutação e deleção α‐.7 foi usado kit comercial Multiplex PCR (QIAGEN®, EUA). A identidade de cada deleção foi obtida pela determinação do tamanho do fragmento amplificado em cada reação. Visto que qualquer uma das deleções remove parte ou todo o gene α‐2 globina, sua amplificação, juntamente com a amplificação de um alelo de deleção, indica que a mutação se encontra em heterozigose. Como controle positivo para o sucesso da amplificação do DNA, foi usado um segmento de 2.350 pb, referente à região 3 não transcrita do gene LIS 1 (fator plaquetário), localizado no cromossomo 17p13.3. Não foram pesquisadas outras mutações, considerando que no Brasil a deleção que leva à α‐Tal é do tipo (3.7).9
As sequências dos iniciadores foram conferidas com as informações disponíveis no NCBI (National Center for Biotechnology Information) com a ferramenta Blast (Basic Local Alignment Search Tool) (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
Além dos fatores genéticos descritos, foi verificada a associação entre HbFetal e AVE. A concentração relativa de hemoglobina fetal no lisado de hemácias foi determinada aos cinco anos e seu valor estratificado, conforme Steinberg,17 em HbFetal ≥ 10% ou HbFetal < 10%.
As crianças e os responsáveis foram previamente esclarecidos sobre a importância deste estudo e autorizaram sua participação com a assinatura do Termo de Consentimento Livre e Esclarecido.
A análise estatística das associações foi feita com aplicação do teste qui‐quadrado, considerando‐se um nível de significância de 5%, no programa SPSS Statistics® 14.0 (IBM Corporation, Somers, NY, EUA).
A pesquisa foi aprovada pelo Comitê de Ética em Pesquisa em 09/10/2009 sob N° 245 e encontra‐se em consonância com o estabelecido na Resolução 466/12 Conselho Nacional de Saúde/MS e com o Código de Ética Médica de 1988 (artigos 122 a 130).
ResultadosDas 110 crianças, 52,7% eram do sexo masculino. A idade média no fim do acompanhamento foi de 11,2 anos, com desvio padrão de 2,84.
Da população do estudo, 60% eram portadores de AF, 33,6% apresentavam genótipo SC e 6,4% eram portadores de S/beta‐Talassemia (duas crianças com S/b0 e cinco com S/b+). A incidência de AF foi de um caso para cada 2.857 crianças nascidas vivas e um caso de DF tipo SC para cada 5.128 nascidos vivos. A renda familiar foi de até dois salários mínimos (SM) em 72,8% dos casos e a escolaridade da mãe, principal cuidadora, atingia o ensino fundamental completo em 60,9%.
A identificação dos haplótipos evidenciou a presença do gene CAR na maioria dos pacientes (89,2%), refletiu a origem da população negra avaliada neste estudo. Houve a presença de genes atípicos, como Senegal e Camarões, anteriormente só descritos na Região Nordeste do Brasil. Não foi possível definir o haplótipo apenas em uma criança. Sete crianças (6,4%) não fizeram o exame genético, devido a recusa ou não comparecimento para coleta de amostra dentro do prazo estipulado para a pesquisa. Das 66 crianças com AF, 89,4% expressaram pelo menos um alelo CAR (tabela 1).
A análise de deleções do gene da α‐globina evidenciou incidência da α‐Tal em 30,9% dos indivíduos. Foram identificados 26,4% de crianças com deleção de um gene e 4,5% com deleção de dois genes. Na nossa população não foram identificadas crianças com deleção de três genes. Em relação à AF, 22,7% das crianças têm a deleção de um gene e 7,6% de dois genes para α‐Tal.
Foram identificadas 19 crianças com AVE, o que representou uma incidência de 17,2%. Dessas, três casos foram identificados por meio da RM feita após manifestação clínica de AVC; 16 casos foram identificados pelo DTC, em 13 dos quais foi evidenciada elevação das velocidades do fluxo cerebral e áreas de isquemia cerebral identificadas na AngioRM cerebral. O aumento da velocidade do fluxo da artéria cerebral média D e sua estenose foram as alterações mais prevalentes (oito casos). A incidência de AVE entre as crianças com AF foi de 27,3%. Não há caso de AVE entre as crianças SC e S/b+. A idade média do primeiro episódio de AVE foi de 7,7 anos, com mínima de seis meses e máxima de 15 anos.
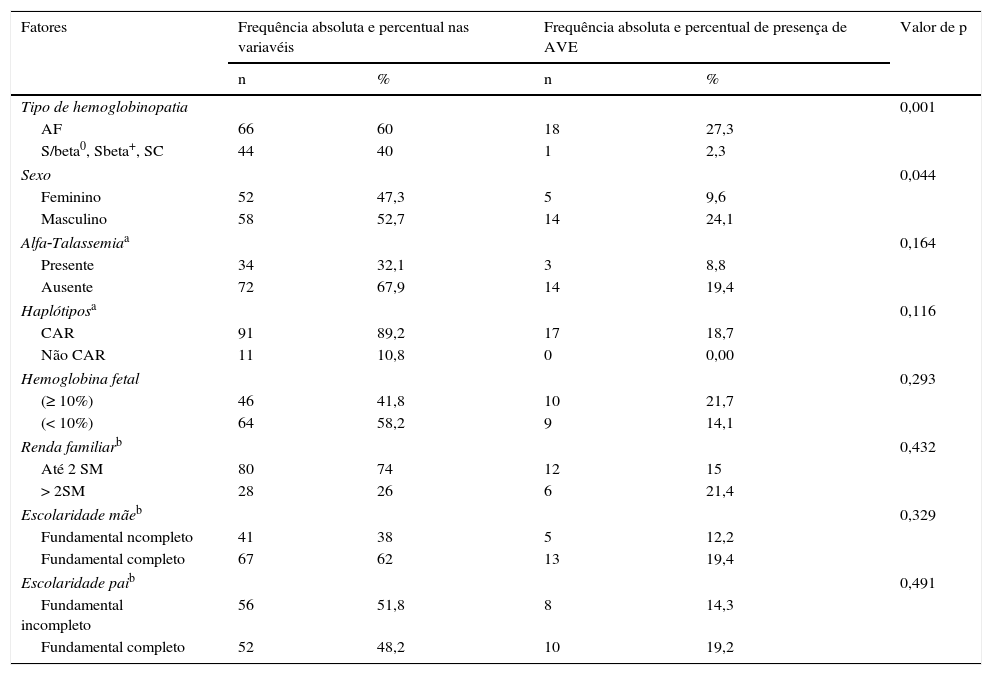
A tabela 2 descreve os fatores associados à presença de AVE. Na amostra analisada, somente uma criança com S/beta‐Talassemia manifestou AVE. Crianças com AF têm 12 vezes mais risco de ter AVE do que as que têm outros tipos de DF (p=0,001). A incidência de AVE entre os meninos foi de 24,1%, contra 9,6% entre as meninas (p=0,044). Os demais fatores não apresentaram associação significante com AVE.
Frequências dos fatores associados à AVE em indivíduos com DF
| Fatores | Frequência absoluta e percentual nas variavéis | Frequência absoluta e percentual de presença de AVE | Valor de p | ||
|---|---|---|---|---|---|
| n | % | n | % | ||
| Tipo de hemoglobinopatia | 0,001 | ||||
| AF | 66 | 60 | 18 | 27,3 | |
| S/beta0, Sbeta+, SC | 44 | 40 | 1 | 2,3 | |
| Sexo | 0,044 | ||||
| Feminino | 52 | 47,3 | 5 | 9,6 | |
| Masculino | 58 | 52,7 | 14 | 24,1 | |
| Alfa‐Talassemiaa | 0,164 | ||||
| Presente | 34 | 32,1 | 3 | 8,8 | |
| Ausente | 72 | 67,9 | 14 | 19,4 | |
| Haplótiposa | 0,116 | ||||
| CAR | 91 | 89,2 | 17 | 18,7 | |
| Não CAR | 11 | 10,8 | 0 | 0,00 | |
| Hemoglobina fetal | 0,293 | ||||
| (≥ 10%) | 46 | 41,8 | 10 | 21,7 | |
| (< 10%) | 64 | 58,2 | 9 | 14,1 | |
| Renda familiarb | 0,432 | ||||
| Até 2 SM | 80 | 74 | 12 | 15 | |
| > 2SM | 28 | 26 | 6 | 21,4 | |
| Escolaridade mãeb | 0,329 | ||||
| Fundamental ncompleto | 41 | 38 | 5 | 12,2 | |
| Fundamental completo | 67 | 62 | 13 | 19,4 | |
| Escolaridade paib | 0,491 | ||||
| Fundamental incompleto | 56 | 51,8 | 8 | 14,3 | |
| Fundamental completo | 52 | 48,2 | 10 | 19,2 | |
AF, anemia falciforme; RCA, República Centro‐Africana; SM, salário mínimo.
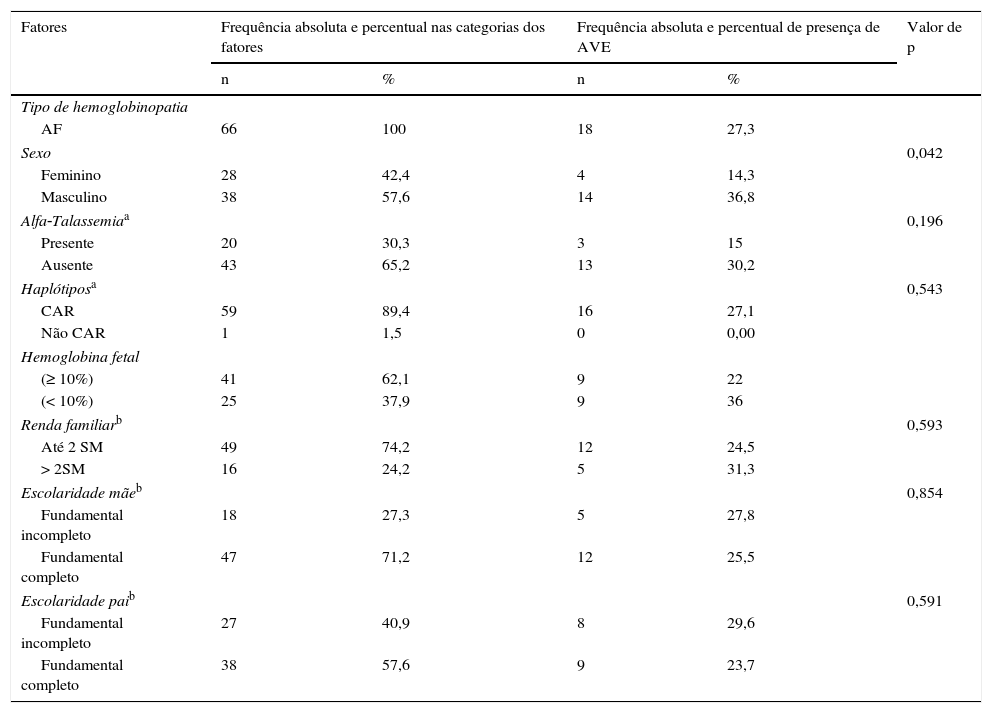
Crianças com AF são mais susceptíveis ao AVE. Por esse motivo, esse grupo foi considerado para verificação de associação de AVE com os demais fatores. Os resultados apresentados na tabela 3 mostram que apenas a variável sexo apresentou associação significante com o desfecho (p=0,042).
Frequências dos fatores associados à AVE em indivíduos com AF
| Fatores | Frequência absoluta e percentual nas categorias dos fatores | Frequência absoluta e percentual de presença de AVE | Valor de p | ||
|---|---|---|---|---|---|
| n | % | n | % | ||
| Tipo de hemoglobinopatia | |||||
| AF | 66 | 100 | 18 | 27,3 | |
| Sexo | 0,042 | ||||
| Feminino | 28 | 42,4 | 4 | 14,3 | |
| Masculino | 38 | 57,6 | 14 | 36,8 | |
| Alfa‐Talassemiaa | 0,196 | ||||
| Presente | 20 | 30,3 | 3 | 15 | |
| Ausente | 43 | 65,2 | 13 | 30,2 | |
| Haplótiposa | 0,543 | ||||
| CAR | 59 | 89,4 | 16 | 27,1 | |
| Não CAR | 1 | 1,5 | 0 | 0,00 | |
| Hemoglobina fetal | |||||
| (≥ 10%) | 41 | 62,1 | 9 | 22 | |
| (< 10%) | 25 | 37,9 | 9 | 36 | |
| Renda familiarb | 0,593 | ||||
| Até 2 SM | 49 | 74,2 | 12 | 24,5 | |
| > 2SM | 16 | 24,2 | 5 | 31,3 | |
| Escolaridade mãeb | 0,854 | ||||
| Fundamental incompleto | 18 | 27,3 | 5 | 27,8 | |
| Fundamental completo | 47 | 71,2 | 12 | 25,5 | |
| Escolaridade paib | 0,591 | ||||
| Fundamental incompleto | 27 | 40,9 | 8 | 29,6 | |
| Fundamental completo | 38 | 57,6 | 9 | 23,7 | |
AF, anemia falciforme; RCA, República Centro‐Africana; SM, salário mínimo.
A complicação mais severa na AF é o AVE, uma das principais causas de morte tanto em crianças quanto em adultos Apesar de ter alta incidência, as causas e os fatores que aumentariam o risco de AVE não são completamente conhecidos. Várias linhas de evidência sugerem que uma assinatura genética poderia influenciar o desenvolvimento de AVE e o efeito combinado desses genes pode afetar a gravidade da DF.2,6,9,15,18,19
Flanangan et al. demonstraram uma associação significativa entre α‐Tal, os polimorfismos genéticos SNPs (single nucleotide polymorphisms) ADCY9 rs2238432 na redução do AVE; três SNPs (ANXA2 rs11853426, TEK rs489347 e TGFBR3 rs284875) foram significantemente associados ao aumento do risco de AVE; a deficiência da glicose 6 fosfato desidrogenase e os haplótipos não foram relacionados ao aumento ou redução do risco. Belisário et al. descreveram aumento do risco de AVE em pacientes que expressam o TNF‐alpha (‐308G>A), não evidenciaram associação significativa com a expressão do polimorfismo VCAM‐1 (c.1238G>C) e associaram a α‐Tal com redução de AVE. Tais resultados sugerem que a associação entre AVE e polimorfismos permanece controversa.1,2,9,15,20‐22 Neste trabalho foram estudadas as associações entre AVE, α‐Tal e haplótipos e não foram encontrados resultados significativos, o que poderia ser decorrente do tamanho reduzido da população do estudo. No nosso estudo não houve relato de AVE entre as crianças portadoras de DF tipo SC e S/beta+ Talassemia, dados consoantes com o Grupo Cooperativo de Estudo em DF.1
Cabe ressaltar que a população desta pesquisa é constituída por todas as crianças diagnosticadas com AF (n=66) na área de abrangência da instituição de saúde que engloba uma população de 730.264 habitantes (Censo IBGE 2010). Sarnaik e Ballas11 enfatizaram a importância de estudos multicêntricos com um grande número de pacientes, com e sem AVE, para determinar a implicação da pesquisa de marcadores genéticos na morbimortalidade na DF.
A incidência de AF, neste estudo, foi de um caso para cada 2.857 nascidos vivos e um caso de DF tipo SC para 5.128. Segundo o Ministério da Saúde, a incidência de DF detectada no PTN foi: Bahia: 1:650, Rio de Janeiro 1:1.300, Pernambuco, Maranhão, Minas Gerais e Goiás: 1:1.400, Espírito Santo 1:1.800, São Paulo 1:4.000, Rio Grande do Sul 1:11.000, Santa Catarina e Paraná 1:13.500.19
Bezerra et al.5 caracterizaram geneticamente uma coorte de 74 crianças com AF em Pernambuco e demonstraram que aproximadamente 65% dos pacientes apresentaram o haplótipo CAR/CAR, uma frequência maior do que as de outros estados na Região Nordeste do Brasil e superior aos 53% encontrados em nosso estudo. Nesta pesquisa não foi identificado o haplótipo Benin/Benin entre as crianças com AF, resultados semelhantes aos encontrados no Rio de Janeiro.13,14 Relatos anteriores mostraram que os haplótipos estão associados com um aumento do risco de AVE.2,11,23,27 De acordo com os nossos dados, a presença do haplótipo CAR (presente em 89,2%) não foi associada ao desenvolvimento de AVE. Nossos resultados são semelhantes aos de Flanangan et al.22 e Loghetto,24 que não encontraram correlação significante entre os haplótipos e AVE. Domingos et al.,12 em 2011, estudaram população de Pernambuco diferente da de Bezerra et al.,5 com 261 pacientes, 67 deles com AVE, e não verificaram associação entre os haplótipos com AVE.
No Brasil, o tipo de deleção que leva à α‐Tal é quase que exclusivamente o do tipo(‐3.7).9 A incidência de α‐Tal encontrada nos indivíduos com AF nesta pesquisa foi de 30,3%, considerando deleção de um e dois genes. Esses dados foram semelhantes aos encontrados em Salvador (28,2%) e superiores aos de Figueiredo, que encontrou 18,8% em São Paulo.4,9
Vários estudos12,15,22 mostraram efeito preventivo de α‐Talassemia no desenvolvimento de AVE em crianças com AF. Belisário et al.9 e Hsu et al.10 relatam que a frequência de α‐Tal foi significativamente maior em pacientes com AF sem alterações ao DTC e sugeriram proteção molecular no desenvolvimento de AVE. Diferentemente, em nossa análise esses marcadores não apresentaram associação significante com risco de AVE, resultados encontrados também por Silva Filho13 e Sommet et al.6 Esses achados sugerem uma possível heterogeneidade genética entre populações, o que poderia justificar as diferenças entre os resultados dos estudos. O manual de conduta e manejo de DF Evidence‐Based Management of Sickle Cell Disease do National Institute of Health (NIH), publicado em 2014, não orienta o rastreio genético para a prevenção de AVE.25
Os fatores socioeconômicos, como renda familiar e escolaridade do cuidador, são descritos como riscos de vulnerabilidade social e determinantes de maiores agravos à saúde da população com DF.3,26‐29 Tais relações não foram observadas neste estudo. Em Minas Gerais, a implantação da política de atenção integral ao paciente com DF propicia um atendimento continuado e multidisciplinar, o que pode ter impactado positivamente e reduzido os efeitos socioeconômicos sobre o manejo da patologia (Centro de Educação e Apoio para Hemoglobinopatias [MG] www.cehmob.org.br).
A incidência de AVE foi significativamente maior nas crianças com AF e no sexo masculino, dado já identificado no International Pediatric Stroke Group.30 A pesquisa da coexistência de presença de α‐Tal e haplótipos neste estudo não evidenciou a correlação na gênese ou na prevenção do AVE. A heterogeneticidade entre as populações previamente avaliadas, a não reprodutibilidade entre estudos e a necessidade de identificação de fatores associados ao AVE em pacientes com AF evidenciam a importância de novas pesquisas, já que essa patologia é a doença monogênica mais prevalente no mundo e uma questão de saúde pública.
FinanciamentoTodo o material de laboratório usado no desenvolvimento do estudo, tais como kits de extração de DNA, reagentes para PCR, enzimas de restrição, tubos e ponteiras, foi adquirido com recursos da Fundação de Amparo à Pesquisa de Minas Gerais (Fapemig) via PPSUS‐ Fapemig (CDS‐APQ‐01431‐10) e do laboratório de pesquisa da Fundação Hemominas. As bolsas de iniciação científica para os estudantes de graduação envolvidos foram financiadas pela Fapemig.
Conflitos de interesseOs autores declaram não haver conflitos de interesse.
Como citar este artigo: Rodrigues DO, Ribeiro LC, Sudário LC, Teixeira MT, Martins ML, Pittella AM, et al. Genetic determinants and stroke in children with sickle cell disease. J Pediatr (Rio J). 2016;92:602–8.
Trabalho desenvolvido na Fundação Hemominas e na Universidade Federal de Juiz de Fora (UFJF), Juiz de Fora, MG, Brasil.
recomendados





